Capítulo 1 Introducción a la Genómica
El presente capítulo es una somera introducción a la genómica y apenas tiene como cometido el asegurarnos de que algunos conceptos básicos sobre la misma, que usaremos durante el curso, se encuentren disponibles. El foco del mismo es en diferentes metodologías para el estudio de los genomas, muchas de las cuales usaremos en distintos ejemplos y secciones del curso. Poco esfuerzo se hace por explicar las bases de la herencia y la estructura y organización de los genomas eucariotas y procariotas, las cuales se asumen conocidas.
Los organismos en nuestro planeta derivan todos de un organismo ancestral, desconocido, y por lo tanto es de esperar que sus genomas compartan determinadas características. Sin embargo, al mismo tiempo podemos observar una enorme diversidad entre los mismos, por lo que resulta fundamental entender los procesos que llevan a esas diferencias. Dichos procesos operan sobre el componente heredable de los organismos, su material genético. El mismo está compuesto por una secuencia ordenada de nucleótidos, por lo cual es común referirse a este coloquialmente como secuencia/s en el ámbito de la genómica y las disciplinas afines. Conocer las secuencias de los genomas de diferentes organismos y compararlas para determinar las diferencias entre estas es, por lo tanto, el primer paso a dar para comprender los procesos que llevaron a dichas diferencias. La secuenciación de genomas es una historia que comenzó en las últimas décadas del siglo pasado y desde ese momento no ha dejado de crecer, en ciertos momentos a velocidad vertiginosa. En particular, la aparición de las denominadas tecnologías de segunda y tercera generación ha permitido pasar de la secuenciación de un genoma por especie a la secuenciación de cientos o miles de individuos de una misma especie. De esta forma, en la acutalidad se está logrando caracterizar mucho mejor la variabilidad biológica existente.
La diversidad existente a nivel de organismos macroscópicos es evidente. El ser humano interacciona con la flora y la fauna desde sus inicios como especie. Entender, preservar e influir sobre esta diversidad biológica es lo que ha permitido en última instancia el desarrollo de las civilizaciones. Resulta intuitivo que enriquecer nuestra comprensión de esta diversidad con aportes desde la genómica pueda resultar valioso. Por otra parte, aunque invisibles los microorganismos juegan un papel relevante en casi todos los ecosistemas a diferentes escalas: desde bosques, praderas, ríos y mares a los ecosistemas internos (e.g., aparato digestivo) y externos (piel) de los organismos. A pesar de su importancia, la diversidad microbiana apenas ha sido explorada, fundamentalmente debido a las dificultades asociadas al cultivo de los microorganismos, muchos de ellos directamente no-cultivables. En este sentido, también la genómica ha jugado un rol destacado en los avances que se han producido en este campo en los últimos años. La metagenómica, la metagenómica funcional y la metatranscriptómica han contribuido a entender la composición de los ecosistemas microbianos, las funciones de los genes involucrados y la regulación de su expresión, respectivamente. Mencionaremos cada una de estas aproximaciones en secciones posteriores de este capítulo.
Aunque a primera vista la informática y la biología puedan parecer áreas de estudio muy alejadas (¿qué se aleja más de la vida que una fría máquina?), veremos que la obtención y procesamiento masivo de datos biológicos han permitido enriquecer de forma importante la perspectiva que tenemos sobre los organismos vivos y la transmisión de información de generación en generación, tanto a nivel individual como poblacional. A lo largo del libro iremos profundizando en esta perspectiva, empleando recursos informáticos para modelar procesos de la genética poblacional y cuantitativa, haciendo un uso enriquecedor de las tecnologías disponibles en la actualidad.
OBJETIVOS DEL CAPÍTULO
\(\square\) Abordar el fenómeno de la variabilidad genética, discutiendo el uso de métricas que nos permiten cuantificar la misma e introduciendo tecnologías que nos permiten la identificación de variantes genéticas en poblaciones.
\(\square\) Obtener una perspectiva de las distintas áreas de estudio de la genómica, incluyendo: - el estudio de la composición nucleotídica de los genomas - el estudio evolutivo de los organismos - el análisis de los patrones de expresión génica
\(\square\) Repasar conceptos básicos de genética y biología molecular, ilustrando en cada caso cómo uso de herramientas estadísticas y computacionales enriquece nuestra perspectiva de los fenómenos biológicos.
1.1 Variabilidad genética
Todos los organismos presentan diferencias observables (fenotípicas) entre ellos. Muchas de estas son producto del desarrollo en determinados ambientes (específicas para cada individuo), mientras que otras se corresponden a las diferencias en el genoma. Excepto los clones (copias idénticas de un mismo individuo), todos los organismos son de alguna manera diferentes entre sí. Aún entre gemelos es posible postular que la epigenética5 puede jugar un rol diferente en su desarrollo. Estas diferencias a nivel genético o genómico son las que nos resultan fundamentales cuando hablamos de mejorar una especie de nuestro interés o en cualquier instancia en la que busquemos seleccionar algunos individuos de una población.
Muchas otras veces estamos más interesados en entender las diferencias entre especies y el rol que juegan las proporciones de individuos de las mismas en la dinámica de un ecosistema. Claramente, la robustez de un ecosistema suele estar asociada al balance de diversas especies, cada una jugando roles diferentes. Pero ese balance no solo depende de la presencia de algún miembro de cada especie, sino de las proporciones que esas especies ocupan en el ecosistema. Tener índices que nos permitan cuantificar y comparar la diversidad existente en diferentes escenarios resulta por lo tanto útil. Tradicionalmente, en ecología se definen tres tipos de medidas de diversidad: la diversidad alfa, la diversidad beta y la diversidad gamma, de acuerdo a la escala del fenómeno estudiado.
Se llama diversidad alfa a la que describe la diversidad de especies dentro de una comunidad6 a una escala pequeña o local. Esta escala es normalmente del tamaño de un ecosistema y es a la que generalmente nos referimos al hablar de una zona.
La diversidad beta describe la diversidad de especies entre dos comunidades o ecosistemas. Involucra una escala mayor, ya que incluye ecosistemas (que correspondían a la diversidad alfa) y en general suele haber alguna distinción geográfica o barrera importante entre las comunidades de referencia.
La diversidad gamma se refiere a una escala de estudio mucho mayor (e.g., un bioma), donde se compara la diversidad de especies entre muchos ecosistemas.
Aunque la diversidad biológica puede cuantificarse de muchas maneras diferentes, los dos factores principales que se tienen en cuenta para medir la diversidad son la riqueza y la uniformidad. La riqueza representa el número de tipos (especies) diferentes entre todos los que identificamos en el área de interés. Sin embargo, nuestra intuición nos dice que la uniformidad representa también una parte importante de la diversidad. Es decir, si en un ambiente tenemos 1 individuo de cada una de 4 especies y 996 individuos de una quinta especie, claramente identificaremos una menor diversidad respecto a otro ambiente que tiene 200 individuos de cada una de las especies. En general, la uniformidad compara la similitud del tamaño de la población de cada una de las especies presentes.
Para cada uno de estos tipos de diversidad se han desarrollado índices que permiten obtener estimaciones de su importancia relativa a otros sistemas. Por ejemplo para la diversidad alfa el índice más sencillo de entender es la entropía de Shannon (llamada así por el matemático Claude Shannon que la propuso para un problema de transmisión de información), que mide la incertidumbre al asignar la identidad de un individuo elegido al azar de la población. Si le llamamos \(p_i\) a la proporción (frecuencia relativa) de individuos de la especie \(i\), de un total de \(k\) especies identificadas en el ecosistema, entonces el índice de Shannon se calcula como
\[\begin{equation} H'=-\sum^k_{i=1} p_i \ln p_i \tag{1.1} \end{equation}\]
Es decir, cada frecuencia es multiplicada por su logaritmo y sumada, con el resultado final multiplicado por \(-1\) ya que cualquiera de los logartitmos será menor o igual a cero (porque para todas las especies \(0 \leqslant p_i \leqslant 1\)). En el caso de una sola especie \(\ln p_i=\ln 1=0\), por lo que la diversidad será cero. Cuanto más homogéneo sea el reparto de las especies, mayor será el índice de Shannon. En particular, el máximo del índice de Shannon se dará cuando \(p_i=\frac{N/k}{N}=\frac{1}{k}\ \forall \ i\), en cuyo caso
\[ \begin{split} H_{max}=-\sum^k_{i=1} \frac{1}{k} \ln \frac{1}{k}=\\ =k\ \frac{1}{k} [\ln 1 -\ln k]=-[0-\ln k] \therefore \\ H_{max}=\ln k \end{split} \tag{1.2} \]
Es decir, el máximo del índice será el logaritmo del número de especies, lo que ocurre cuando todas las especies tienen el mismo número de integrantes. Podemos ver entonces que el índice de Shannon no sólo nos permite comparar la diversidad biológica de forma cuantitativa, sino que además nos permite predecir bajo qué condiciones ésta se maximiza. Lo que es más, esta predicción se corresponde con nuestra intuición de que una mayor diversidad debe reflejar un reparto más equitativo de los individuos para las diferentes especies (concepto de uniformidad mencionado anteriormente).
Resulta claro que los valores que se pueden obtener para el índice de Shannon dependerán del número de especies presentes (\(k\)) en el ecosistema estudiado. En este sentido, resulta útil un índice relacionado directamente al índice de Shannon es el propuesto por Pielou (1969), que estandariza el índice de Shannon por la riqueza, lo que lo deja como un índice de uniformidad (“evenness”, en inglés) que se calcular como
\[\begin{equation} E=\frac{H'}{H_{max}}=\frac{H'}{\ln k} \tag{1.3} \end{equation}\]
Así, es posible tener una noción más clara respecto a la diversidad alfa presente en el ecosistema estudiado: cuanto más se acerca este índice \(E\) a 1, más cerca se estará del máximo teórico según el índice de Shannon.
Otro índice popular es el índice de Simpson. El índice de Simpson (\(D\)) mide la probabilidad de que dos individuos seleccionados al azar de una muestra pertenezcan a la misma especie o categoría. Es decir, si \(n_i\) representa el número de individuos de la especie o categoría \(i\) y \(N\) el número total de individuos, es decir \(N=\sum_i n_i\), entonces
\[\begin{equation} D=\sum^k_{i=1} \frac{n_i(n_i-1)}{N(N-1)} \tag{1.4} \end{equation}\]
La intuición de este índice es que si los individuos se concentran en pocos tipos diferentes el valor de \(D\) será más alto, es decir lo contrario a lo que esperaríamos para un índice de diversidad. De hecho, con este índice 0 representa una diversidad infinita y 1, ninguna diversidad. Es decir, cuanto mayor sea el valor de \(D\), menor será la diversidad. Para superar este problema, se suele restar a 1 el valor de \(D\), es decir se reporta \(1-D\). Otra alternativa es reportar el recíproco de \(D\), es decir \(1/D\). Cuando el número de individuos es muy grande, entonces en general \(\frac{n_i -1}{N-1} \to \frac{n_i}{N}\), por lo que
\[ \begin{split} D=\sum^k_{i=1} \frac{n_i(n_i-1)}{N(N-1)}=\sum^k_{i=1} \frac{n_i}{N}\frac{(n_i-1)}{(N-1)} \approx \sum^k_{i=1} \frac{n_i}{N}\frac{(n_i)}{(N)} \therefore \\ D \approx \sum^k_{i=1} \frac{n^2_i}{N^2} \end{split} \tag{1.5} \]
Notemos que hasta el momento no se han tenido en cuenta posibles errores al momento de determinar el número de especies k presentes en una muestra (e.g. por falta de muestreo de organismos de una especie presente en el ecosistema en números muy bajos).
El índice de cobertura de Good es usado a veces como estimador de diversidad alfa y se calcula mediante la siguiente ecuación:
\[\begin{equation} C=1-\frac{F_1}{N} \tag{1.6} \end{equation}\]
donde \(F_1\) es el número de OTUs (“Operational Taxonomic Units”; Unidades Taxonómicas Operacionales, en inglés) en la muestra con un solo representante. OTU es una forma de expresar una unidad taxónomica particular a un nivel de la taxonomía determinado. Por ejemplo, en algunos casos las OTUs pueden representar especies, pero otras veces representan niveles más altos o más bajos de la taxonomía (de ahí el término “operacional”). La idea del índice es capturar de alguna manera la probabilidad de perder OTUs debido a un tema de muestreo. Es decir, aquellas OTUs que no fueron secuenciadas pero que se encontraban presentes, las cuales se hallarían de haberse realizado muestreos con tamaños muestrales mucho mayores.
La diversidad juega un papel fundamental en la resiliencia de los sistemas, tanto la diversidad entre especies como dentro de especies. Por ejemplo, la pérdida de diversidad asociada a la revolución verde entre la década del 60 y del 80 del siglo XX representa una pérdida irrecuperable de patrimonio genético desarrollado durante miles y millones de años. Lamentablemente, es poco probable que la biotecnología pueda recuperar en breve lo que la naturaleza desarrolló durante millones de años, por lo que resulta trascendental pensar en conservar la diversidad en nuestro planeta.
Ejemplo 1.1
Luego de un muestreo realizado a partir de metagenómica en dos ambientes diferentes, la distribución de especies (OTUs) en los mismos es la siguiente:
| Especie | Ambiente.1 | Ambiente.2 |
|---|---|---|
| OTU 1 | \(60\) | \(1\) |
| OTU 2 | \(150\) | \(1\) |
| OTU 3 | \(0\) | \(297\) |
| OTU 4 | \(390\) | \(1\) |
| OTU 5 | \(400\) | \(700\) |
| Total | \(1000\) | \(1000\) |
Calcular los difentes índices para los dos ambientes y discutir los resultados.
Como el total de “individuos” (reads) obtenidos en ambas muestras es \(1000\), alcanza con dividir entre ese número para tener la frecuencia relativa. Por lo tanto, utilizando la ecuación (1.1) el índice de Shannon para el primer ambiente es igual a
\[ \begin{split} H'*1=-\sum^k*{i=1} p_i \ln p_i =\\ =-[0,06 \times \ln 0,06 + 0,15 \times \ln 0,15 + 0,39 \times \ln 0,39 +\\ - 0,4 \times \ln 0,4] \therefore \\ H'_1=1,1871 \end{split} \]
mientras que para el segundo ambiente el mismo es
\[ \begin{split} H'*2=-\sum^k*{i=1} p_i \ln p_i =\\ =-[0,001 \times \ln 0,001 + 0,001 \times \ln 0,001 + 0,297 \times \ln 0,297 +\\ +0,001 \times \ln 0,001 + 0,7 \times \ln 0,7] \therefore \\ H'_2=0,63096 \end{split} \]
Notar que en el Ambiente 1 no se encontró la especie OTU 3, por lo que no se incluye en los cálculos (de hecho \(\ln 0=-\infty\)). De acuerdo con esto, según el índice de Shannon el Ambiente 1 es más diverso que el Ambiente 2. Para medir la uniformidad utilizando el índice propuesto por Pielou (1969), primero tenemos que calcular el máximo posible para el índice de Shannon, que a partir de la ecuación (1.2) sabemos que es igual al logaritmo del número de OTUs, es decir \(H_{max_1}=\ln k=\ln 4=1,3863\) y \(H_{max_2}=\ln k=\ln 5=1,6094\). Por lo tanto, aplicando la ecuación (1.3), tenemos que el índice de uniformidad para los dos ambientes será
\[ \begin{split} E_1=\frac{H'*1}{H*{max_1}}=\frac{1,1871}{1,3863}= 0,8563 \\ E_2=\frac{H'*2}{H*{max_2}}=\frac{0,63096}{1,6094}= 0,3920 \end{split} \]
lo que habla de una uniformidad mucho menor en el Ambiente 2.
Para el índice de Simpson vamos a utilizar la ecuación (1.4), por lo que para el Ambiente 1 el mismo será
\[ \begin{split} D_1=\sum^k_{i=1} \frac{n_i(n_i-1)}{N(N-1)}=\\ =\left[\frac{60(59)+150(149)+390(389)+400(399)}{1000 \times 999}\right]=0,3375 \end{split} \]
mientras que para el Ambiente 2 su valor será
\[ \begin{split} D_2=\sum^k_{i=1} \frac{n_i(n_i-1)}{N(N-1)}=\\ =\left[\frac{1(0)+1(0)+297(296)+1(0)+700(699)}{1000 \times 999}\right]=0,5778 \end{split} \]
Si utilizamos el criterio de reportar \(1-D\), entonces \(1-D_1=1-0,6625\), mientras que \(1-D_2=1-0,4222\). Si utilizamos el criterio del recíproco, entonces \(1/D_1=2,9626\) y \(1/D_2=1,7307\), por lo que se aprecia claramente que la diversidad en el Ambiente 1 es mayor que la del Ambiente 2 de acuerdo a este índice.
Finalmente, de acuerdo a la ecuación (1.6), teniendo en cuenta que en el primer ambiente no hay ningún OTU con un único representante mientras que en el segundo hay 3, el índice de cobertura de Good para los dos ambientes sería
\[ \begin{split} C_1=1-\frac{0}{1000}=1 \\ C_2=1-\frac{3}{1000}=0,997 \end{split} \]
PARA RECORDAR
Tradicionalmente, en ecología se definen tres tipos de medidas de diversidad de acuerdo a la escala del fenómeno estudiado:
- Se llama diversidad alfa a la que describe la diversidad de especies dentro de una comunidad a una escala pequeña o local. Esta escala es normalmente del tamaño de un ecosistema y es a la que generalmente nos referimos al hablar de una zona.
- La diversidad beta describe la diversidad de especies entre dos comunidades o ecosistemas. Involucra una escala mayor, ya que incluye ecosistemas (que correspondían a la diversidad alfa) y en general suele haber alguna distinción geográfica o barrera importante entre las comunidades de referencia.
- La diversidad gamma se refiere a una escala de estudio mucho mayor (e.g.,un bioma), donde se compara la diversidad de especies entre muchos ecosistemas.
Aunque la diversidad biológica puede cuantificarse de muchas maneras diferentes, los dos factores principales que se tienen en cuenta para medir la diversidad son la riqueza y la uniformidad. La riqueza representa el número de tipos (especies) diferentes entre todos los que identificamos en el área de interés, mientras que la uniformidad compara la similitud del tamaño de la población de cada una de las especies presentes.
Para la diversidad alfa, el índice más sencillo de entender es la entropía de Shannon, que mide la incertidumbre al asignar la identidad de un individuo elegido al azar de la población. Si le llamamos \(p_i\) a la proporción (frecuencia relativa) de individuos de la especie \(i\), de un total de \(k\) especies identificadas en el ecosistema, entonces el índice de Shannon se calcula como: \(H'=-\sum^k_{i=1} p_i \ln p_i\). Cuanto más homogéneo sea el reparto de las especies, mayor será el índice de Shannon. El máximo del mismo será cuando todas las especies tienen el mismo número de integrantes.
El índice de Simpson (\(D\)) mide la probabilidad de que dos individuos seleccionados al azar de una muestra pertenezcan a la misma especie o categoría. Es decir, si \(n_i\) representa el número de individuos de la especie o categoría \(i\) y \(N\) el número total de individuos (i.e., \(N=\sum_i n_i\)), entonces \(D=\sum^k_{i=1} \frac{n_i(n_i-1)}{N(N-1)}\). La intuición de este índice es que si los individuos se concentran en pocos tipos diferentes el valor de \(D\) será más alto, al contrario de lo que esperaríamos para un índice de diversidad. Para superar este problema, se suele restar a 1 el valor de \(D\) (se reporta \(1-D\)).
1.1.1 Detectando la variabilidad: diferentes técnicas
En las últimas décadas han ganado una enorme relevancia un conjunto de disciplinas con base en avances tecnológicos que permiten la obtención masiva de información biológica global para una condición dada. Esto se logra a través de la automatización de diversos protocolos de bioquímica y biología molecular, tales como la secuenciación de nucleótidos o péptidos provenientes de una muestra biológica. Estas disciplinas son identificables por la utilización del sufijo “-ómica” en su nombre. La genómica es una de estas disciplinas, focalizándose en el estudio del genoma de diferentes organismos a través de las tecnologías de secuenciación masiva. Existen también la transcriptomica (estudio global de los transcriptos expresados por un organismo o conjunto de células), la proteómica (estudio de las proteńas expresadas en una condición dada) o la metabolómica (estudio del perfil metabólico), entre otros. Este enfoque de análisis también se puede aplicar para estudiar varios organismos a la vez (e.g. al tomarse muestras ambientales), surgiendo así un subconjunto de discplinas “meta-”. A modo de ejemplo, la metagenómica se encarga del ensamblado y estudio de genomas microbianos en muestras provenientes de diferentes lugares. En la Sección Metagenómica y metatranscriptómica se detalla un esquema típico de trabajo en metagenómica. Como es de esperar, estas técnicas de obtención masiva de datos han impactado de forma notable nuestro entendimiento de la variabilidad genética entre organismos.
Para entender la variabilidad genética es necesario determinar algún marcador genético cuyas variantes podamos detectar en muchos individuos (e.v., obteniendo las secuencias para el marcador en esta muestra). Un ejemplo de estudios iniciales de diversidad genética es la utilización de grupos sanguíneos, el cual veremos en el próximo capítulo. Hasta hace algunos años la búsqueda de marcadores fiables de diversidad genética representaba un enorme desafío, y se desarrollaron una plétora de métodos para obtener marcadores moleculares de diversidad. Tanto el trabajo como los costos eran enormes, lo que dificultaba el desarrollo de estudios a gran escala en este sentido. Afortunadamente, a fines del siglo XX las técnicas de secuenciación masiva habían avanzado lo suficiente como para incluir centenas de individuos en los estudios. Más aún, el desarrollo de técnicas como los microarrays posibilitaron la automatización de muchos análisis (en especial los de expresión), pero también resultaron fundamentales a la hora de abaratar los costos para estudios masivos de diversidad.
Los microarrays son un arreglo de sondas moleculares colocadas en una placa. Originalmente contenían apenas unas decenas de sondas; la robotización y la aplicación de tecnologías que se usan en la fabricación de semiconductores permitieron alcanzar densidades del orden del millón de sondas por array (placa). Una sonda molecular, en este caso, es una secuencia específica para un gen (o una región de un gen) y que es, idealmente, única en el genoma de referencia. Esta sonda se encuentra fija en la placa, y la hibridación del ADN o el ADN copia (en los estudios de transcriptómica) de un individuo es acoplada a algún tipo de señal (e.g., emisión de luz en determinada frecuencia al ser iluminada por un láser). La presencia de señal permite se asocia, por lo tanto, a la presencia del ADN de interés en la muestra proveniente del individuo.
Esta tecnología permite, por ejemplo, identificar si la secuencia presente en el organismo es de un tipo u otro en el caso de los SNPs (polimorfismos de un solo nucleótido). En el caso de las especies diploides, es posible identificar el genotipo para un organismo. Para conocer la diversidad del genoma humano es usual utilizar chips de densidad media (10-60K de SNPs) o de alta densidad (hasta un millón de SNPs). En especies de interés comercial existen diferentes tipos de alternativas, desde arrays de baja densidad (apenas unos pocos miles de SNPs), media, hasta los de alta densidad, como el BovineHD que comprende más de 777 mil SNPs, compatible con todas las razas bovinas lecheras o de carne.
A pesar del enorme éxito de los microarrays para estudiar la diversidad genética en diversas poblaciones de diferentes especies, los mismos padecen de un problema fundamental: para crear una sonda para medir variabilidad hay que conocer las secuencias de regiones que contengan variabilidad. Es decir, para estudiar la variabilidad mediante esta técnica hay que tener alguna información previa de qué regiones son variables. Esto se debe a que la suma de los tamaños de las sondas son extremadamente pequeños en relación al largo total de los genomas. Por ejemplo, en genoma humano un chip de alta densidad cubre 1 millón de posiciones de las más de 3.000 millones que tiene el genoma (~0.0003%). Para elegir bien ese millón de posiciones hay que tener muy buena información previa: si se eligen posiciones que casi no varíen en la población, se obtendrá muy poca información sobre la variabilidad genética en las poblaciones estudiadas. Además, otro problema derivado de los microarrays y de este sesgo en los marcadores a incluir es que luego de diseñado el array solo va a captar variabilidad en las posiciones del genoma para el que fue diseñado. Si una nueva población aparece con variabilidad importante en otras regiones, el array no va a ser capaz de detectarlo. Por ejemplo, en genoma humano, hasta hace poco tiempo los arrays poseían un importante sesgo “eurocéntrico”, porque la mayor parte de la información previa venía de Europa. Esto dejaba fuera de los mismos sitios de alta variabilidad en poblaciones asiáticas o amerindias, por ejemplo. Estudios que utilizaran dichos arrays terminarían determinando que estas poblaciones tenían menos variabilidad genética que la real.
Afortunadamente, las tecnologías de secuenciación masiva que se desarrollaron a comienzo del siglo XXI cambiaron definitivamente el panorama, ya que permiten obtener las secuencias de decenas o cientos de individuos a precio y tiempos razonables. Como referencia, el esfuerzo global para obtener la secuencia del primer genoma humano llevó más de 10 años a decenas de laboratorios repartidos en el mundo, y costó más de 3.000 millones de dólares. Actualmente, un genoma humano tarda en secuenciarse menos de una semana (el record mundial, establecido recientemente, es de 5 horas y 2 minutos) y cuesta en promedio unos mil dólares (sin incluir los costos del análisis posterior). Más aún, si bien las tecnologías de segunda generación (Roche 454, Illumina y SOLiD) tuvieron un éxito enorme, la flexibilidad de las mismas era algo limitada y en particular era muy difícil reducir los tiempos de secuenciación, además de tener ciertos sesgos específicos en los resultados. En la segunda década de este siglo comenzaron a aparecer y consolidarse otras nuevas tecnologías, que se dieron en llamar de tercera generación y que incluyen los secuenciadores IonTorrent, los de Oxford Nanopore Technologies y los PacBio. En conjunto, estas tecnologías, sumadas a Illumina que es la única de las de segunda generación que sobrevive con mucho éxito, permiten tener una flexibilidad enorme en tamaños y costos de equipamientos y reactivos, en sesgos y distribución de errores, así como en la velocidad de secuenciación.
1.1.2 Metagenómica y metatranscriptómica
En muchos casos nos interesa determinar la estructura de alguna comunidad biológica y estudiar su evolución en el tiempo, o simplemente comparar esa estructura en diferentes condiciones. Por ejemplo, la microbiota ruminal es fundamental para los procesos que determinan la degradación de la materia orgánica que el animal recibe, estando asociada también a qué productos de esta degradación recibirá el animal y en qué proporciones. ¿Qué variables afectan más la composición de la microbiota? ¿El pH ruminal? ¿El porcentaje de materia seca? ¿La proporción de fibra? En el plano de la salud, ¿qué microorganismos se encuentran presentes en la ubre sana y qué microorganismos en la glándula de un animal con mastitis? ¿La infección es a causa de un microorganismo dominante o el patógeno no precisa ser el que domine el ecosistema ruminal? ¿El rol del patógeno es directo sobre la salud del animal o su rol se limita a desestabilizar la ecología de la glándula?
Para poder responder a cualquiera de estas preguntas es necesario poder caracterizar la comunidad microbiana presente. A diferencia de lo que ocurre con especies de plantas, hongos y animales que generalmente podemos percibir y contar a simple vista, con los microorganismos nos ocurre que necesitamos identificarlos de formas más indirectas. Si bien usando el microscopio (con algo de suerte) podríamos ser capaces de reconocer el género de algunas bacterias presentes en una muestra, difícilmente seamos capaces de reconocer a la mayoría. Seríamos mucho menos capaces de reconocer las especies y cepas de estas bacterias, y muchísimo menos capaces de cuantificar la proporción de cada grupo bacteriano. Haciendo un paralelismo, para entender la comunidad vegetal de un bosque no alcanza con determinar la presencia de al menos un ejemplar de una especie: es fundamental entender la proporción que dicha especie ocupa en el ecosistema.
Como ya mencionamos, el hecho de que todos los organismos en nuestro planeta “desciendan” del mismo organismo ancestral, a veces conocido como LUCA (“Last Unknown Common Ancestor”, en inglés), nos sugiere que sería de esperar encontrar ciertas regularidades en la información genética de los mismos. Es decir, diferentes formas de vida son el producto de modificaciones a formas de vida preexistentes, al menos hasta llegar a LUCA, por lo que de alguna manera todos los seres vivos comparten ciertas características. Las diferencias entre especies, como sabemos, se corresponden en gran medida a cambios en el genoma de los mismos, tanto a nivel de mutaciones puntuales como de inserciones y deleciones, duplicaciones de regiones del genoma o aún re-arreglos cromosómicos. La clave para identificar especies es ubicar secuencias lo suficientemente conservadas como para que deban encontrarse en todos los organismos de interés, pero al mismo tiempo con suficientes cambios entre especies como para poder identificarlas en forma unívoca.
Dentro de las secuencias con estas características, la maquinaria vinculada a la traducción de la información es ciertamente privilegiada ya que constituye un elemento clave para cualquier forma de vida: la capacidad de traducir la información genética en los efectores, que suelen ser la proteínas. La traducción de la información implica una maquinaria tan compleja que resulta completamente implausible su surgimiento de forma independiente en diferentes linajes de la vida, más aún dado que cualquier especie ya hubiese precisado de una para existir. Además, debido a su enorme complejidad se trata de una maquinaria muy ajustada y donde es posible introducir muy pocas variaciones sin que la maquinaria deje de funcionar completamente, en particular porque el fitness7 de los organismos tiene, en muchos casos, una vinculación directa con la tasa de producción de las proteínas. Un caso de particular importancia es el ARN ribosomal (ARNr). Este tipo de marcador molecular está constituido por una serie de moléculas de ARN que dependen de su estructura espacial para poder cumplir su rol. Esto incluye apareamientos Watson-Crick entre regiones complementarias, pero también bases modificadas post-transcripcionalmente para asegurar la estabilidad de la molécula y la fidelidad de la traducción.
En procariotas, el ARNr 16S es una molécula usada en forma extensiva para identificar el genero y especie de los organismos. De hecho, la propuesta para un sistema de clasificación de la vida de Woese, Kandler, and Wheelis (1990), representada en la Figura 1.1, se basa en las diferencias observadas en la molécula de 16S y su equivalente en eucariotas, el ARNr 18S. De acuerdo con esta forma de organizar las diferentes especies, existen tres grandes dominios de la vida: Bacteria, Archaea y Eukarya.8. Es importante notar que a diferencia de la taxonomía basada en similitud morfológica, la clasificación propuesta por Woese, Kandler, and Wheelis (1990) se basa en similitud en las secuencias genómicas, donde de acuerdo a determinados modelos se pueden asignar relaciones de proximidad y distancia entre secuencias pertenecientes a diferentes organismos. En particular, la reconstrucción filogenética se realiza generalmente a partir de secuencias de genes ortólogos9, en este caso el ARN ribosomal 16S, por ejemplo. Si bien desde el punto de vista morfológico los miembros del dominio Archaea no son, aparentemente, muy distintos de los miembros de Bacteria, desde el punto de vista evolutivo poseen grandes diferencias que los hacen acreedores de su propio dominio.
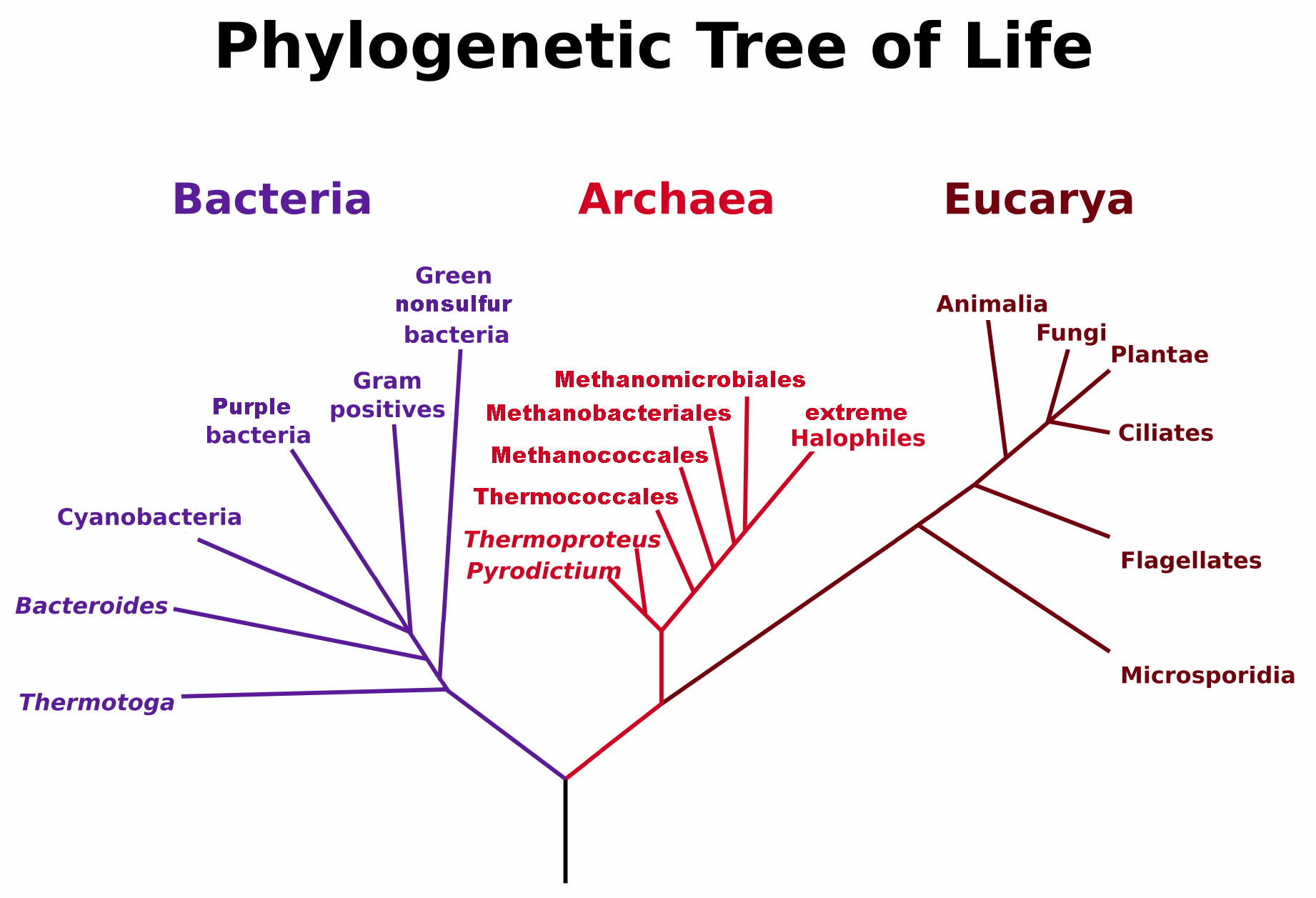
Figura 1.1: Árbol filogenético de los 3 dominios biológicos, Bacteria, Archaea y Eukarya. Con una barra vertical se representa la existencia de LUCA, el último ancestro común, desconocido. (Por Maulucioni - Trabajo Propio, CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=24740337).
Las técnicas de secuenciación masiva implicaron un salto enorme para la caracterízación de poblaciones microbianas, ya que permiten la secuenciación de miles y hasta millones de fragmentos de secuencias en muy poco tiempo y en forma paralela. La idea más básica consiste en secuenciar una determinada secuencia o región que se encuentre en todos los organismos de interés y que presente suficiente variabilidad, como es el caso del ARNr 16S. Esquemáticamente, los pasos serían los siguientes:
- Determinar el grupo de organismos de interés, así como la molécula y región a secuenciar. Las regiones hipervariables V3 y V4 del ARNr 16S son buenas candidatas iniciales para procariotas. El gen tiene una longitud aproximada de 1.500 pb y contiene nueve regiones variables intercaladas entre regiones conservadas. Qué región del ARNr 16S secuenciar es un área de debate, y la misma puede variar dependiendo de cosas como los objetivos experimentales, el diseño y el tipo de muestra, aunque las regiones V3 y V4 son ampliamente utilizadas.
- Extraer los ácidos nucleicos de interés y amplificar por PCR las regiones de interés. Los protocolos permiten en general utilizar “barcodes”, que son secuencias específicas que se agregan a las secuencias (un “barcode” para cada muestra), lo que permite correr en paralelo hasta 96 muestras en una misma “corrida” del secuenciador.
- Secuenciar las muestras preparadas de acuerdo al protocolo. Existen diferentes tecnologías y en distintos tamaños, lo que permite secuenciar desde unas poquitas muestras hasta centenas de ellas a un costo razonable y en tiempos razonables. Las distintas tecnologías poseen problemas diferentes, pero en general no introducen sesgos importantes en la composición estimada de la muestra.
- Control de calidad y limpieza de las secuencias obtenidas. Es una serie de procesos a nivel bioinformático que tienen por objetivo eliminar las secuencias de los adaptadores que se agregaron durante la preparación de las bibliotecas, separar las secuencias de acuerdo al correspondiente “barcode” y calcular una serie de parámetros que indican la calidad de la corrida y de cada una de las muestras.
- Agrupamiento de secuencias y mapeo contra bases de datos con secuencias identificadas. Se trata del corazón de la idea y consiste en que cada secuencia o fragmento producido durante la secuenciación debe tener uno idéntico depositado en una base de datos con secuencias de organismos. Hay diferentes estrategias respecto a cómo agrupar los fragmentos antes de “mapearlos” contra las bases de datos y diferentes bases de datos de organismos, pero esto ya es materia específica de la bioinformática.
- Una vez mapeadas las secuencias obtenidas, es decir que a cada una le asignamos un taxón determinado (puede llegar a ser una especie, aunque a veces la identificación queda a un nivel más alto, por ejemplo género), entonces alcanza con contar cuántas veces aparece cada organismo en una muestra (es decir, cuántos fragmentos secuenciados mapean al mismo taxón). Esto nos permite organizar la información en una tabla que puede tener las muestras en filas y los taxa en las columnas.
- Análisis de la tabla de conteos. Por lo general la tabla generada de conteos tiene diferencias importantes en el número de secuencias obtenidas por muestra ya que no se obtiene la misma cantidad de ácidos nucléicos en cada muestra a pesar de los esfuerzos. Para la parte de exploración suelen usarse análisis exploratorios multivariados, como el escalado multidimensional (MDS), una técnica con cierta relación al PCA en su versión más clásica (que se conoce como Principal Coordinates Analysis, PCoA). La idea es reducir la dimensionalidad del problema de tal forma que se pueda representar en pocas dimensiones con poca pérdida de información. En el mejor de los casos, después es posible correlacionar directamente las posiciones de la muestras en las principales dimensiones con factores externos, tales como el pH, la temperatura, ubicación geográfica o cualquier otro factor que sea relevante. En la Figura 1.2 se observa la representación gráfica de los dos primeros ejes en un análisis MDS realizado como control de calidad en un experimento de transcriptómica RNA-seq con Leptospira biflexa. Se observa que las muestras se agrupan de acuerdo a la condición biológica y que los dos factores relevantes desde el punto de vista biológico (fase de crecimiento y tiempo) son las que separan las muestras entre sí.
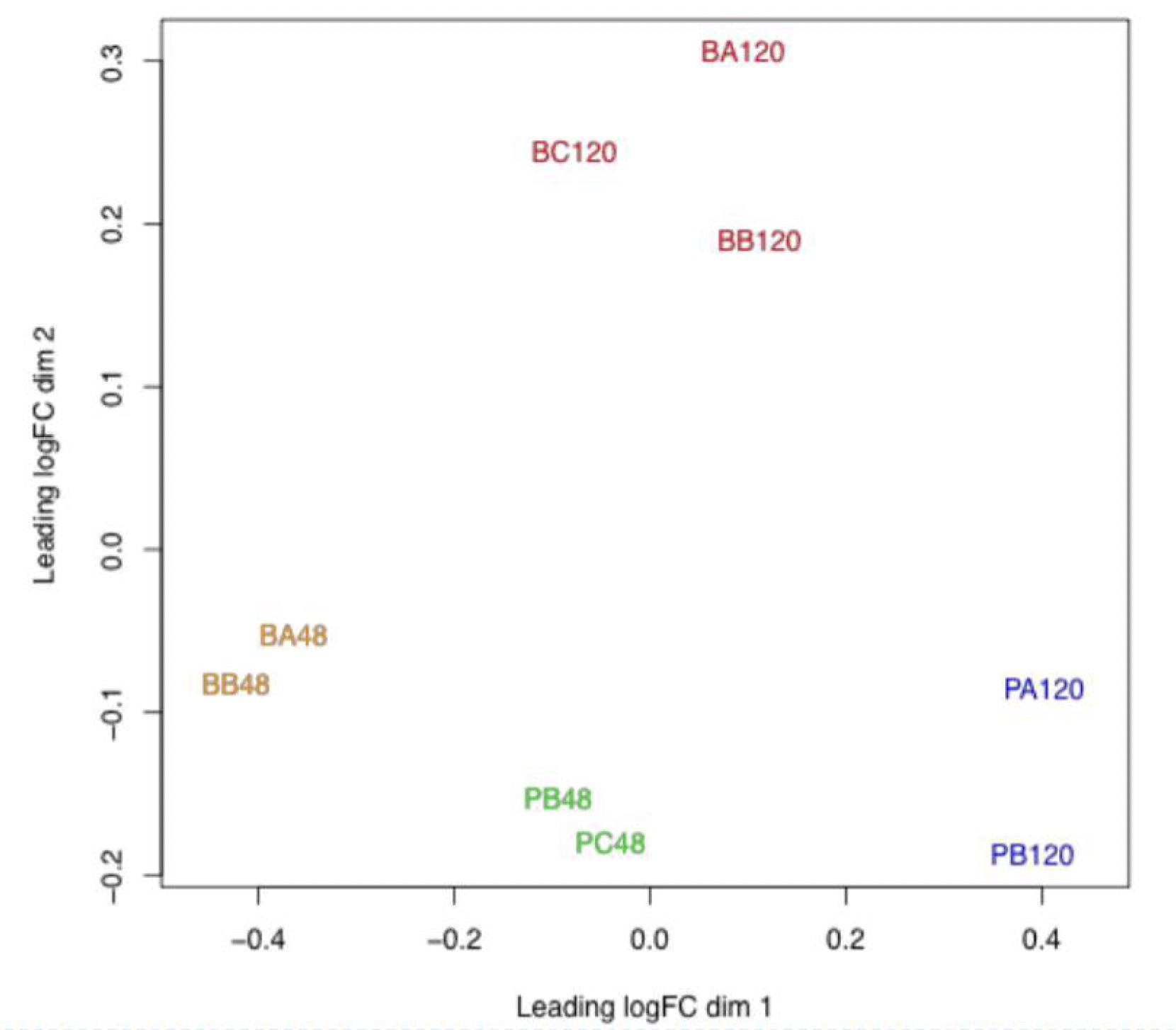
Figura 1.2: Escalado multidimensional en un experimento de transcriptómica de Leptospira biflexa. El experimento incluía dos factores, la fase de crecimiento (biofilm y planctónica) y dos tiempos (48 y 120 horas), en 3 réplicas (A, B y C) por condición. Alguna de las réplicas fue descartada por motivos de calidad (figura tomada de Iraola et al. (2016)).
Con las tecnologías de segunda generación (Roche 454, Illumina, SOLiD) era y aún es común secuenciar fragmentos, como vimos recién. Sin embargo, con tecnologías de tercera generación como la de Oxford Nanopore Technologies es común secuenciar toda la molécula de 16S ya que el largo de la lectura no está determinado a priori (como sí en Illumina) y es normal obtener reads (lecturas del secuenciador) de miles o aún decenas de miles de pares de bases. Por otro lado, existen otras formas de metagenómica que no pasan por la secuenciación de una región o de una secuencia en particular. En metagenómica funcional la idea es secuenciar todo el ADN presente en la muestra, en lugar de determinadas secuencias en particular. Esto permite capturar no solo la variabilidad de organismos presentes en la muestra sino que también identificar secuencias de ellos que puedan ser relevantes al problema biológico en cuestión.
Por ejemplo, si un determinado microorganismo se encuentra asociado a un determinado estado patológico de un sistema, conocer las secuencias de genes del mismo puede ser relevante para interpretar los mecanismos de acción del mismo. Asumiendo que el organismo no se encuentre completamente secuenciado y disponible en las bases de datos públicos, la función de las secuencias relevantes se puede deducir a partir del mapeo contra las bases de datos de secuencias anotadas (por ejemplo GenBank), usando BLAST. Por otro lado, si varios organismos están expresando las mismas vías, por ejemplo a causa del exceso de un determinado producto o metabolito en el medio, entonces aparecerá una plétora de secuencias relacionadas por la función, pero provenientes de diferentes organismos.
Un paso más adelante se encuentra la metatranscriptómica que tiene por objetivo la caracterización de todo el ARN mensajero (ARNm) presente en cada muestra. La diferencia más importante con la genómica funcional es que mientras que en esta última cada genoma aporta lo mismo y por lo tanto el factor de ponderación es la proporción de cada taxa (v.g. especie) en la muestra, en la metatranscriptómica existe la misma ponderación, pero también la ponderación por expresión de las secuencias (en realidad un balance entre la tasa de transcripción y la de decaimiento). De esta forma, más allá de identificar qué secuencias están presentes en la muestra, podemos cuantificar (en forma relativa) la “importancia” de cada una. Por ejemplo, en situaciones de organismos patógenos, si el mismo está expresando genes de resistencia a antibióticos, podemos identificar las secuencias responsables de esto a través de la metatranscriptómica.
Las aplicaciones de todas estas técnicas, metagenómica, metagenómica funcional y metatranscriptómica han permitido avanzar en varios campos donde entender el funcionamiento del ecosistema microbiológicos es fundamental. Por ejemplo, en agricultura resulta muy claro que desde el punto de vista de las plantas el suelo no es solamente el sustrato y las sustancias químicas disponibles para las mismas; se trata de un ecosistema complejo donde los microorganismos juegan un rol fundamental, tanto los de vida libre como los asociados a las plantas. En este sentido, resulta fundamental entender la diversidad de microorganismos en los distintos suelos, su evolución a lo largo del tiempo y cómo eso se relaciona con la productividad del suelo.
Ejemplos en el área de producción animal también son ubicuos. El rumen es un órgano complejo cuya principal misión consiste en la fermentación de los alimentos ingeridos y la preparación para la posterior absorción de nutrientes a nivel intestinal. Se trata de un ambiente esencialmente anaerobio (ausencia de oxígeno) donde proliferan múltiples microorganismos, entre los que se destacan bacterias, protozoos, arqueas metanogénicas, hongos y virus. La mayor parte de los microorganismos presentes nunca han sido cultivados, en parte por la dificultad de cultivar anaerobios, pero las técnicas de metagenómica han permitido enormes avances al respecto. Claramente, la microbiota ruminal es dinámica y suele verse afectada por diversos factores, que incluyen la dieta, cambios en la fisiología del animal producto de la preñez, cambios ambientales o la estación del año. Los distintos tipos de bacterias que actuan en el rumen (de acuerdo a su función en la digestión) comprenden las celulolíticas, lactolíticas, sacarolíticas, lipolíticas, amilolíticas y metanogénicas ureolíticas, entre otras. Entender cómo las distintas variables afectan la composición de la microbiota ruminal es esencial para predecir los impactos en la salud animal y en la producción.
En salud animal como en salud vegetal resulta fundamental entender cuáles son los patógenos causantes de una enfermedad. En general, muchos de los microoorganismos que se encuentran en el órgano afectado son desconocidos y en muchos casos no son cultivables. Como las condiciones de cultivo suelen ser fundamentales en la bacteriología clásica, existe un sesgo importante en asignar las causas de la patogenicidad a organismos cultivables. Esto nos deja con una parte de la historia conocida y otra parte (posiblemente igual de relevante) sin conocer, ya que en muchos casos el origen de una patología puede ser el producto de un desarreglo en la comunidad que lleve a patógenos oportunistas a tomar la oportunidad. Más aún, uno de los temas de mayor preocupación para la salud mundial, en el paradigma de Una Sola Salud (One Health, en inglés) es la resistencia a antibióticos que se ha desarrollado a escala global y que amenaza dejarnos sin tratamiento ante enfermedades que parecían superadas. Frente a esto parece fundamental empezar a trabajar el tema desde una perspectiva racional, tanto a nivel de las políticas y la investigación como a nivel del trabajo de campo. Para un manejo racional es fundamental, a su vez, conocer los organismos causantes de cada patología específica, así como los potenciales mecanismos de resistencia desarrollados, ya que el uso infructuoso de antibióticos no hace otra cosa que aumentar las posibilidades de desarrollar otras resistencias.
En este último sentido, la metagenómica puede constituirse en una herramienta muy poderosa para la invetigación, por ejemplo en el estudio de la mastitis bovina, analizando los cambios en la flora microbiana de la glándula mamaria en animales sanos cuando pasan a un estado de mastitis. En principio, no solo es posible estudiar la composición microbiana durante la evolución de la enfermedad sino que mediante metagenómica funcional, y especialmente mediante metatranscriptómica, se puede analizar el resistoma (el conjunto de los genes relacionados a la resistencia) de la ubre (Hoque et al. (2020)). Además, otras técnicas masivas que ya se están utilizando en forma rutinaria en el diagnóstico de enfermedades infecciosas en humano, como la espectrometría de masas MALDI-TOF10, prometen revolucionar la velocidad y precisión de diagnóstico en animales de producción, así como mejorar la predicción de resistencias antes de aplicar antibióticos (Nonnemann et al. (2019)).
Ejemplo 1.2
Interpretar la Figura 1.2 sabiendo que la letra inicial (“P” de planctónica, es decir vida libre, o “B” de biofilm) representa la fase del crecimiento de colonias de la bacteria Leptopsira interrogans, la segunda letra representa la réplica biológica del experimento (“A”, “B” o “C”, para tres réplicas) y los número finales representan las horas a las que se extrajo la muestra a partir del inicio del experimento.
En general, en todos los experimentos biológicos donde queremos generalizar las conclusiones a la población debemos incluir una muestra representativa de individuos a comparar. Los experimentos de transcriptómica son lo suficientemente complejos y caros como para que el número de réplicas no sea elevado (3 por condición, en este caso).
Originalmente se trata de un experimento factorial balanceado del tipos 2x2, ya que tenemos dos factores (la fase de crecimiento -vida libre o biofilm- y los tiempos -48 y 120 horas-), cada uno con dos niveles; en cada celda del diseño factorial tenemos 3 réplicas. Se trata de un experimento de RNA-seq (transcriptómica), por lo que los datos que originan el gráfico salen de una tabla de conteo de “reads”.
Lo primero que se observar en la figura es que algunas muestras fueron descartadas del análisis, de acuerdo con los autores debido a que no cumplían los requisitos de calidad previamente establecidos. Lo segundo que se observa es que las muestras se agrupan razonablemente de acuerdo a la condición biológica, es decir, cada celda del diseño factorial (que están representadas por distintos colores). Seguramente, las muestras no consideradas caerían lejos de su grupo correspondiente (lo que no es criterio para descartarlas), pero también lejos de de todas las otras muestras.
Finalmente, la distribución de las muestras no sigue el patrón de que cada celda del diseño factorial (cada color en el gráfico), ocupe un cuadrante diferente, en particular cada eje representando uno de los factores. Entonces, podemos deducir que la relación entre los factores y las fuentes de varianción no es totalmente ortogonal (i.e., no son totalmente independientes). Sin embargo, es posible trazar una línea que divida a las de vida libre (planctónicas) de las sésiles (biofilm) y otra línea que separe las de 48 hs vs las de 120 horas (de hecho, infinitas de estas rectas en ambos casos), por lo que parece existir “buena biología” en las causas de la separación.
PARA RECORDAR
- Para entender la variabilidad genética es necesario conocer las secuencias, o al menos algunas secuencias que nos puedan servir de marcador, en muchos individuos. Los microarrays son un arreglo de sondas moleculares colocadas en una placa.
- Una sonda molecular, en este caso, es una secuencia específica a un gen o una región de un gen y que es (idealmente) única en el genoma de referencia. Cuando el ADN o el ADN copia (en los estudios de transcriptómica) de un individuo se hibrida con dicha sonda (que está fijada en la placa) se produce algún tipo de señal, por ejemplo la emisión de luz en determinada frecuencia al ser iluminada por un laser. Esta tecnología permite, por ejemplo, identificar si la secuencia presente en el organismo es de un tipo u otro en el caso de los SNPs (polimor- fismos de un solo nucleótido) e identificar el genotipo en el caso de las especies diploides.
- Es importante tener en cuenta que para crear una sonda para medir variabilidad hay que conocer de antemano las secuencias de regiones que contengan variabilidad, y que el array solo va a captar variabilidad en las posiciones del genoma para el que fue diseñado.
- La metatranscriptómica tiene por objetivo la caracterización de todo el ARN mensajero (ARNm) presente en cada muestra. La diferencia más importante con la genómica funcional es que mientras que en esta última cada genoma aporta lo mismo y por lo tanto el factor de ponderación es la proporción de cada taxa (v.g., especie) en la muestra, en la metatranscriptómica existe la misma ponderación, pero también la ponderación por expresión de las secuencias. De esta forma, más allá de identificar qué secuencias están presentes en la muestra, podemos cuantificar (en forma relativa) la “importancia” de cada una.
1.2 Genómica composicional
Si bien todos los organismos derivan del mismo organismo ancestral (LUCA) y por lo tanto deben compartir ciertas características en sus genomas, los mismos evolucionan y son la base de las diferencias entre los distintos organismos. A nivel genómico la evolución se da por un conjunto de eventos como las mutaciones puntuales, las duplicaciones de genes y regiones, o las inserciones y “deleciones” de secuencias nucleotídicas. Claramente, la mayor parte de estos eventos producen cambios importantes a nivel composicional de los genomas. Por ejemplo, las mutaciones pueden llevar a una acumulación de cambios completamente al azar en los genomas, o puede tener determinados tipos de sesgos que favorezcan que las mutaciones sean hacia determinadas bases (por ejemplo, mutaciones desde las bases A y T hacia las base G y C; recordar que en el ADN doble cadena A se aparea con T y C con G). Los eventos de duplicación génica, por otro lado, generan cierta libertad para que ambas evolucionen más libremente, ya que en principio una de las dos secuencias sigue siendo funcional (hasta que también deje de serlo) y de esta forma se pueden explorar nuevas funciones para una de las dos secuencias.
En general, el conjunto de estos cambios hace que resulte interesante para entender las bases de la evolución en ambientes particulares, o aún en general, el conocimiento de las secuencias de los diferentes organismos y la comparación de las mismas. El estudio de las variaciones en la composición de los genomas es lo que se puede llamar genómica composicional y forma parte de la genómica comparativa. A continuación veremos el tema de la variación en composición, tanto desde el punto de vista de los nucleótidos, como de los codones en las secuencias codificantes y finalmente de los aminoácidos que forman las proteínas.
Contenido GC genómico, génico, correlaciones y \(\text{GC}_{\text{skew}}\)
La proporción de bases guanina y citosina (G y C, respectivamente) que componen una secuencia se conoce como el contenido GC de la misma, o G+C, o aún GC% (cuando expresado en porcentaje)11. Por las reglas de apareamiento de bases (A-T y G-C), no tiene importancia en la determinación de dicho contenido cual hebra del ADN se considera para el cálculo de esta proporción.
Reglas de Chargaff
- Primera regla de paridad La primera regla sostiene que una molécula de ADN de doble cadena tiene a nivel global las siguientes igualdades en el porcentaje de bases:%A=%T y %G=%C. La validación rigurosa de la norma constituye la base del apareamiento de Watson-Crick de la doble hélice del ADN.
- Segunda regla de paridad La segunda regla es que tanto %A∼%T y %G∼%C son válidos para cada una de las dos hebras de ADN. Esto describe solamente una caracterísstica global de la composición de bases en una sola hebra de ADN.
En principio, uno podría preguntarse cuál es la importancia de medir esta proporción en particular y no otra de las tres posibles combinaciones de dos bases distintas12. Es más, alguna de ellas resultan obviamente relevantes, como la proporción de purinas vs. pirimidinas (AG vs. CT). Entonces, ¿qué es lo que hace tan relevante al contenido GC de las secuencias?
Transiciones y transversiones
- Los cambios entre PURINAS (A y G) o entre PIRIMIDINAS (C y T) se llaman transiciones.
- Los cambios de PURINAS a PRIDIMINAS, o al revés, se llaman transversiones.
- Las transiciones son mucho más frecuentes que las transversiones.
Existen varias razones, como veremos más adelante, pero desde el punto de vista bioquímico tal vez la más obvia sea la diferencia en el número de puentes de hidrógeno entre los apareamientos A-T y los G-C, dos para el primero y tres para el segundo, así como diferencias en el “stacking” resultante13. Esto conduce directamente a importantes diferencias en las propiedades físicas de la doble hebra. Por ejemplo, existe un incremento en el “melting point” (temperatura en la cual la mitad de las dobles hélices de ADN se desnaturalizan) en función del contenido GC (Yakovchuk, Protozanova, and Frank-Kamenetskii 2006). Pero más importante aún, en secuencias codificantes el código genético (tanto el “universal” como todas sus variantes) establece un mapeo entre la secuencia de ADN y la proteína que será traducida a partir de la misma. Dada la estructura “redundante” del código genético, algunos aminoácidos presentan poca dependencia del contenido GC, mientras que otros se encuentran fuertemente asociados a este problema (ya que los codones correspondientes tienen un sesgo importante).
En la Figura 1.3 se aprecia la tabla correspondiente al código genético universal14.
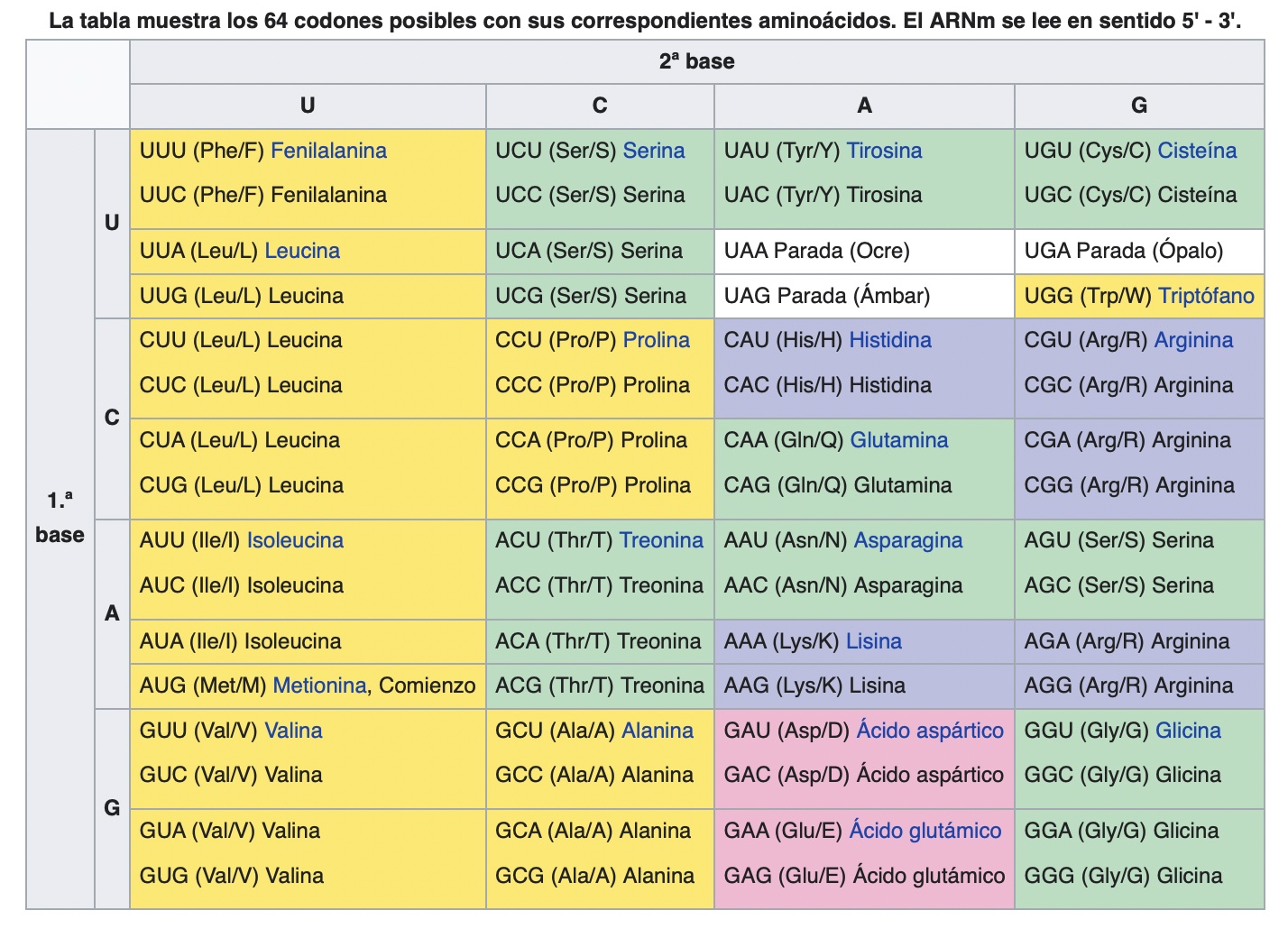
Figura 1.3: Tabla del código genético universal. En amarillo los aminoácidos apolares, en verdes los polares, azul básicos y rosa ácidos, con blanco correspondiendo a los codones de parada (de Wikipedia, https://es.wikipedia.org/wiki/Código_genético).
Claramente, de acuerdo a los colores, que representan propiedades de los aminoácidos, los mismos se encuentran distribuidos en forma bastante organizada de acuerdo a estas propiedades. Cada columna marca una base diferente en segunda posición del codón (U, C, A, G). Por ejemplo, la tercera columna incluye los codones con segunda base A (adenina) y la misma codifica para aminoácidos mayormente codificados por duetos (i.e.,por dos codones). La primera columna corresponde a codones con segunda base U (uracilo, correspondiente en ADN a T) y codifica para aminoácidos apolares.
Alguna propiedades del código genético universal
- 2 Singletons (Met y Trp), 9 Duetos (Phe, Tyr, His, Gln, Asn, Lys, Asp, Glu, Cys), 1 Terceto (Ile), 5 Cuartetos (Val, Pro, Thr, Ala, Gly) y 3 Sextetos (Leu, Ser, Arg).
- La tercera columna (A en la segunda base) codifica duetos: 2/3 de los cambios en tercera base no son sinónimos.
- La primera base del codón es muy conservada: un cambio de bloque-fila implica en general cambio de aminoácido, excepto en sextetos Arg y Leu.
- La segunda base del codón es la más conservada: un cambio de columna implica un cambio de aminoácido.
- La tercera base es la más neutra y por lo tanto menos conservada (rol de los cuartetos). Los cambios de aminoácidos (de cuál a cuál) no son todos equivalentes en su impacto: hay cambios conservativos y otros disruptivos.
Entre los aminoácidos codificados por codones con un fuerte sesgo GC(AT) tenemos: Phe (5 A/T en 6 posiciones), Ile (8 A/T en 9) y Tyr (5 A/T en 6). ¿Son utilizados estos aminoácidos de forma homogénea en los diferentes genes codificantes de los organismos?. Veamos por ejemplo cómo es el uso de estos aminoácidos en el genoma de Buchnera aphidicola str. Cc (Cinara cedri), una bacteria parásita intracelular obligada con contenido GC de 20.1%. Mientras que en la proteína codificada por la secuencia con mayor contenido GC del genoma (36.7%) el uso de estos 3 aminoácidos corresponde al 16%, en la proteína con menor contenido GC (8.6%) el uso de los mismos sube a 48%. Además, nos podemos preguntar, ¿las diferencias en contenido GC a nivel genómico pueden dar lugar a diferencias en el contenido GC de las secuencias codificantes?. ¿Esto puede llevar a diferencias entre organismos con genomas con contenido GC diferente?. En principio, podemos pensar que estas diferencias no tendrían de por sí una consecuencia necesaria a nivel de diferencias entre organismos, ya que de hecho el contenido GC podría ser una simple consecuencia de los aminoácidos constituyentes de las proteínas codificadas en el organismo y no al revés. Es decir, la distribución del contenido GC de las proteínas de un organismo podría ser solo el reflejo de la constitución de las mismas.
Sin embargo, de ser esto cierto esperaríamos que organismos con una
composición relativamente similar a nivel de sus proteínas
constituyentes tuvieran similar contenido GC, y esto no es lo que ocurre.
De hecho, es bien conocido que entre los organismos procariotas
(Bacteria y Archaea) el contenido GC genómico varía desde 25% a 75%
aproximadamente (Sueoka 1962), con algunos organismos aún más extremos. Estas variaciones marcadas se dan incluso entre organismos de un mismo filo. Más aún, la
bacteria Anaeromyxobacter dehalogenans 2CP-C, primer cultivo puro de
una myxobacteria capaz de crecer en forma anaeróbica, con un contenido
GC genómico de 74.9%, está constituída por proteínas cuyo contenido GC
va entre 56.5% y 88.0%. Comparando esto con los datos de B. aphidicola
mencionados anteriormente, observamos que no existe superposición. Es decir, ambas bacterias no
tendrían ninguna proteína de similar composición. Sabemos, sin embargo,
que ambas comparten un origen evolutivo único, y por lo tanto descienden
de un mismo organismo. Además, ambas cuentan con una serie de proteínas
con idéntica función, pero de hecho con la composición aminoacídica de estas es muy
diferente. Claramente, estos hechos van en contra de la línea de razonamiento planteada anteriormente.
¿Qué es entonces lo que nos permite pensar que sería el contenido GC el que conduce el proceso de sustitución de aminoácidos? Hay varios motivos para pensar en este sentido. El primero es que los procariotas, cuyo genoma es en su mayoría codificante, poseen una importante homogeneidad composicional (restringida por la necesidad de usar todos los aminoácidos). Esto se manifiesta aún en las regiones intergénicas, que no tienen mayores restricciones composicionales. Un ejemplo del alto nivel de homogeneidad se puede ver al comparar los intervalos inter-cuartílicos de contenido GC codificante, que tanto en B. aphidicola como en A. dehalogenans es de 6%; en otras palabras, la mitad de las proteínas dentro de cada bacteria difieren en menos de 6% de contenido GC (comparar contra más de 50% de diferencia entre el contenido GC del genoma de ellas).
Pero, además, existe otra evidencia fuerte. Así como calculamos el contenido GC de una
secuencia, también podemos calcular el contenido GC en cada una de las
posiciones de los codones de la misma. Como hay tres posiciones por
codón tenemos entonces \(GC_1\), \(GC_2\) y \(GC_3\), de acuerdo al contenido
GC solo considerando una posición a la vez. Si observamos el código
genético con detenimiento vamos a observar que la tercera posición es la
“más sinónima”, es decir, la posición en la que una mayor proporción de
los cambios de base no afectan el aminoácido codificado. De hecho,
excepto Met y Trp, todos los aminoácidos poseen codones
sinónimos, con opción entre G/C y A/T. De acuerdo con esto, si el
contenido GC no estuviera conduciendo el proceso de sustitución de
aminoácidos, se podría esperar que el \(GC_3\) se mantuviera variando
libremente entre los distintos genomas, con casi tanta varianza entre
como dentro de genomas. Sin embargo esto no es así, ya que existe una
fuerte correlación positiva entre el contenido GC y el \(GC_3\), tanto a
nivel de secuencias como considerando los genomas enteros (en
procariotas) y aún con las regiones intergénicas flanqueantes
(Zerial et al. 1986).
Además de los sesgos en contenido GC que discutimos más arriba, existen otros sesgos menos obvios pero con muy importantes consecuencias prácticas. Por ejemplo, si consideramos una región particular de la mayoría de los genomas bacterianos es posible observar una diferencia importante (dentro de cada hebra) entre el número de bases G y C, así como entre A y T. De hecho, este fenómeno se encuentra asociado a la replicación de los genomas bacterianos (usualmente circulares) y típicamente la hebra “leading” (“líder”, en inglés) se encuentra enriquecida en G y T, mientras que la “lagging” (“rezagada”, en inglés) se encuentra enriquecida en C y A. Las desviaciones de las frecuencias G=C y A=T se conocen como \(\text{GC}*{\text{skew}}\) y \(\text{AT}*{\text{skew}}\), respectivamente 15. Una aplicación práctica evidente de esto es la determinación (aproximada) del origen de replicación, ya que en este (y en la posición opuesta dentro del genoma circular) habrá un cambio de signo del \(\text{GC}*{\text{skew}}\) (asociado a la densidad de genes en una hebra y otra). Una forma de visualizar esto es graficando el \(\text{GC}*{\text{skew}}\) en las ordenadas contra la posición del genoma (centro de la ventana) a partir del cual fue calculado, identificando el cambio de signo en las ordenadas. Otra alternativa, generalmente más efectiva, es plotear el \(\text{GC}_{\text{skew}}\) acumulado contra la posición y observar los máximos y mínimos del gráfico, que usualmente están a una distancia de medio genoma (Grigoriev 1998).
Si bien existe una fuerte controversia sobre la explicación causal de
todas estas correlaciones, así como de correlaciones entre el contenido
GC y factores del ambiente donde viven los organismos, resulta claro que el contenido GC
tiene un importante rol en la evolución de los organismos y en los
procesos moleculares que rigen a los mismos (incluso se debe tener en
cuenta a la hora de calcular algunos índices genómicos o en los procesos
bioquímicos que requieran la denaturalización del ADN). Existen diversas
herramientas para el cálculo del contenido GC, alguna de ellas web
(v.gr., Mobyle http://mobyle.pasteur.fr/) que sirven para un cálculo
rápido de una o pocas secuencias. Sin embargo, si el cálculo involucra
muchas secuencias lo usual es recurrir a la programación de tipo
“scripting”, usualmente en lenguajes como Python
(http://python.org/), Perl (http://www.perl.org/), o Java
(http://www.java.com; todos ellos poseen módulos “Bio” que facilitan
todas las tareas), en R (http://cran.r-project.org/) a través de
bibliotecas como seqinr, en bash (cuando se trabaja en Linux-UNIX o
Mac OS-X), o usando paquetes como EMBOSS
(http://emboss.sourceforge.net/). Si se desea trabajar con genomas
procariotas ya secuenciados, una página interesante es la de OligoWeb
(http://insilico.ehu.es/oligoweb/), ya que además de las frecuencias de
nucleótidos de los distintos genomas ya tiene calculados otros índices y
estadísticos.
Uso de codones
Habíamos hablado antes de que los genomas procariotas eran extremadamente amplios en el rango de contenidos G+C y que, dado lo reducido de lo no-codificante, estas variaciones debían tener implicancias en el uso de codones. Es más, si comparamos entre genomas veremos que existe una clara correlación lineal entre el contenido GC del genoma con \(GC_1\), \(GC_2\) y \(GC_3\), aunque con diferentes pendientes. En general \(GC_3\) es una posición sinónima, es decir al cambiar de base en ella no hay cambio de aminoácido (debido a la degeneración del código genético; ver los cuartetos por ejemplo). \(GC_1\) y \(GC_2\) son mucho más restringidas en su variación, dado que suelen implicar un cambio de aminoácido, lo que por lo general afecta negativamente la función de la proteína donde sucede dicho cambio.
Esta redundancia en el código genético, sumado a las posibilidades de
reconocimiento alternativo codón/anticodón (no-Watson-Crick), permiten la
evolución en los genomas de diferentes estrategias de uso de codones
(UC) (Eduardo P. C. Rocha 2004). Esto, que puede verse entre genomas, también tiene su
correlato a nivel interno en muchos genomas. Es conocido que en algunos
genomas procariotas los genes altamente expresados tienden a usar
codones mayores, que suelen ser reconocidos en forma de apareamiento Watson-Crick por los tRNAs más abundantes, aunque hay distintos modelos (ver la discusión en
(Eduardo P. C. Rocha 2004)). Lograr comprender los factores que inciden en el uso de
codones (también puede pensarse para aminoácidos) suele requerir de
alguna forma de “resumir” la información.
En un genoma procariota promedio tenemos unos 4000 genes y los codones de aminoácidos con degeneración son 59 (los 64 menos los 3 stop, menos Met y Trp, que son codificados por un único codón cada uno), lo que se puede resumir en una tabla de esas dimensiones. Utilizando la misma, podríamos analizar si distintos genes utilizan de forma diferente los codones, e hipotetizar cuál puede ser el motivo subyacente a este fenómeno. Para quitar la influencia del uso de aminoácidos como factor que influye en esta perspectiva (cada proteína tiene un uso diferencial de AAs), se puede estandarizar a la interna de cada aminoácido. Una de las estandarizaciones posibles es el RSCU (Sharp, Tuohy, and Mosurski 1986), en el que el número observado de codones para un aminoácido es dividido por el número esperado si el uso dentro de un aminoácido fuera equiprobable. Es decir,
\[\begin{equation} \text{RSCU}*{ij}=\frac{x*{ij}}{\frac{1}{n_i}\sum_{j=1}^{n_i}\ x_{ij}} \tag{1.7} \end{equation}\]
donde \(x_{ij}\) es el número de ocurrencias del codón \(j\) para el aminoácido \(i\) y \(n_i\) es la multiplicidad del aminoácido \(i\) (es decir, el número de codones que lo codifican).
De cualquier forma, aún luego de estandarizar seguimos teniendo la tabla de 4000x59 (i.e. cada gen en una fila, y el índice RSCU calculado para cada uno de los 59 codones considerados). La mayor parte de los humanos tenemos una limitada capacidad para visualizar datos en espacios de más de 3 dimensiones, por lo que necesitamos reducir la “dimensionalidad” del problema. Podríamos pensar en representar las 59 variables originales de a pares (\(\binom{59}{2}=59*58/2=1711\)), o aún de a tres (\(\binom{59}{3}=59*58*57/6=32509\)) para ver interacciones entre más variables, pero estos números hacen imposible poder extraer conclusiones relevantes en forma sistemática. Afortunadamente, existen diferentes técnicas estadísticas exploratorias multivariadas que nos permiten abordar este tipo de cuestiones.
Probablemente, de las técnicas usuales de análisis exploratorio multivariado, la más conocidas es el Análisis de Componentes Principales (PCA, del inglés Principal Component Analysis), aunque existen otros miembros prominentes de la familia. Uno de los más interesantes para nuestro tipo de problema es el Análisis de Correspondencia (COA, ver Sección Exploración multivariada: Análisis de Correspondencia). El mismo nos permite reducir la variación en las 59 variables originales a unas pocas dimensiones (cada una de ellas una combinación de las originales) que capturan la mayor parte de la variabilidad. De esta forma, el problema pasa a ser identificar qué variables biológicas se asocian a cada una de estas dimensiones.
Ejemplos típicos de variables biológicas asociadas a la variación en el uso de codones dentro de genomas procariotas son el contenido GC del gen, su nivel de expresión, la hidropatía promedio de la proteína, la hebra en que se encuentra el gen, el \(\text{GC}_{\text{skew}}\) y la precisión en la traducción (Ermolaeva 2001). Diferencias en el uso de codones entre genomas han sido reportadas asociadas a varios factores eco-fisiológicos, entre ellos la temperatura óptima de crecimiento y adaptaciones a hiper-salinidad. Un programa diseñado específicamente para el análisis composicional es el codonW (http://codonw.sourceforge.net/), que analiza las frecuencias de bases, codones y aminoácidos, calcula diferentes índices estadísticos, así como permite realizar un COA en codones y AAs.
Existen diversos índices, calculados a partir de las frecuencias de codones, que permiten identificar sesgos importantes en UC, que pueden ser suficientemente indicativos de por sí, o combinados con la información del COA. Por ejemplo, el Nc (número efectivo de codones) es un índice del uso de codones intragénico. Puede tomar valores extremos de 20, cuando un solo codón por aminoácido es utilizado en ese gen, a 61, cuando hay una equi-distribución de los mismos (F. Wright 1990). Este índice es sensible al contenido GC del genoma, y una mejora fue sugerida posteriormente que toma en cuenta este factor (Novembre 2002). Otros dos índices, “codon bias index” (CBI) y “frequency of optimal codons” (Fop), calculan el sesgo a partir de un juego de codones “óptimos”, derivados respectivamente de un conjunto particular de genes o de la concentración de los tRNAs. Una aproximación totalmente diferente, que no depende de un conjunto particular de genes, es el “Codon Adaptation Index” (CAI)(Sharp and Li 1987), que ha demostrado ser un indicador muy razonable del nivel de expresión de los genes (basado en comparaciones con técnicas para medir experimentalmente la expresión génica, como los microarrays). El CAI es simplemente la media geométrica de los RSCU relativos (al máximo RSCU para ese AA) de todos los codones de una proteína. Con la misma notación que más arriba, si consideramos \(\text{w}*{ij}=RSCU*{ij}/RSCU_{i,max}\), entonces se define \(\text{CAI}=\exp({\frac{1}{L}\sum_{k=1}^L\ln \text{w}_{ij}})\).
Pese a los buenos resultados del CAI, el mismo presenta también importantes debilidades, como la no-linealidad y una sobre-estimación de las desviaciones para secuencias cortas. En el 2009, Roymondal y colaboradores (Roymondal, Das, and Sahoo 2009) proponen una aproximación muy diferente, basada en las frecuencias de cada base en las distintas posiciones de los codones, que lleva al índice RCB. Estos autores proponen para cada codón xyz calcular el \(\text{RCB}\) de acuerdo a la fórmula:
\[\begin{equation} \text{RCB}_{xyz}=\frac{f(xyz)}{f_1(x)f_2(y)f_3(z)} \tag{1.8} \end{equation}\]
(donde \(f_1(x)\), \(f_2(y)\), \(f_3(z)\) son las frecuencias de las bases \(x\), \(y\) y \(z\) en las posiciones 1, 2 y 3 respectivamente), para luego resumir toda la información a través de la media geométrica, \(RCB=(\prod_{l=1}^LRCB_{xyz}(l))^{1/L}-1\). Este nuevo índice presenta ventajas respecto al CAI, pero aún presenta un problema importante para secuencias cortas, lo que llevó a sugerir una modificación del mismo (Relative Codon Adaptation, RCA), basada en el uso de pseudo-conteos (Fox and Erill 2010).
Un índice de expresión tiene mucha utilidad en la exploración de los factores que influyen en la composición diferente de los genes, pero un punto interesante es la posible relación entre la expresión y el efecto de “gene dosage” (más copias de genes disponibles a medida que la replicación avanza), resultado de favorecer los genes altamente expresados en una posición cercana al origen de replicación. Para investigar este efecto basta con ver cual es la relación entre el CAI y la posición. Una forma fácil consiste en un análogo del \(GC_{skew}\) acumulado, calculados como desvíos del CAI promedio y compararlo con las posiciones genómicas. Si existe un efecto de “gene dosage” el patrón de CAI debería mostrar un pico en la zona del origen de replicación.
Un soporte adicional a la hora de entender lo que ocurre en el uso de codones cuando comparamos dos grupos, por ejemplo genes de alta expresión con los de baja expresión, es a través de las tablas de uso de codones. Las mismas contienen el conteo total por codón para cada grupo y si las agrupamos por AA podemos calcular la probabilidad de que las diferencias en uso de codones (para cada codón) se deban al azar. Para esto, dentro de cada AA se contruye una tabla de 2x2 (2 grupos a comparar y codón a analizar vs la suma de los otros dentro de AA) y con la misma realiza un test de \(\chi^2\) con un grado de libertad. Es importante tener en cuenta las múltiples comparaciones a realizar (el número de hipótesis a ensayar; v.gr., el número de codones) a la hora de definir el umbral de significancia para corregir adecuadamente.
Finalmente, el uso de codones de un organismo puede tener implicancias directas de carácter tecnológico. Como vimos más arriba, dentro de las mayores fuentes de variación en UC se encuentra en algunos organismos la expresión, con codones muy usados en los genes de alta expresión y un uso más plano en proteínas de baja expresión. Esto influye inclusive en el plegamiento de las proteínas, cambiando la solubilidad de la misma (Cortazzo et al. 2002). Cuando deseamos expresar proteínas recombinantes resulta muy importante considerar no solo la secuencia de AAs de la proteína a expresar, sino también que el UC se adapte al UC en genes de alta expresión en el organismo hospedero.
Ejemplo 1.3
Teniendo en cuenta todas las secuencias codificantes, en una bacteria de interés biotecnológico se observó el siguiente conteo de codones para dos aminoácidos:
| Aminoácido | Codón | Conteo |
|---|---|---|
| Arg (R) | CGU | \(666\) |
| Arg (R) | CGC | \(1320\) |
| Arg (R) | CGA | \(1457\) |
| Arg (R) | CGG | \(1818\) |
| Arg (R) | AGA | \(5913\) |
| Arg (R) | AGG | \(5120\) |
| Val (V) | GUU | \(7123\) |
| Val (V) | GUC | \(2144\) |
| Val (V) | GUA | \(1125\) |
| Val (V) | GUG | \(8213\) |
Calcular el RSCU para cada codón y determinar cuáles son los usados y si hay diferencias entre los aminoácidos en la forma en que usan los codones.
De acuerdo a la ecuación (1.7), el RSCU se calcula como
\[\begin{equation} RSCU_{ij}=\frac{x_{ij}}{\frac{1}{n_i}\sum_{j=1}^{n_i}\ x_{ij}} \end{equation}\]
donde \(x_{ij}\) es el número de ocurrencias del codón \(j\) para el aminoácido \(i\). Tenemos dos aminoácidos diferentes, por lo vamos a calcular el RSCU de cada codón dentro de cada aminoácido. Lo primero es obtener la suma de codones en cada aminoácido, por lo que tenemos que para Arginina es igual a \(666+1320+1457+1818+5913+5120=16294\), mientras que para Valina es de \(7123+2144+1125+8213=18605\). Este es el número total de codones para cada uno de los aminoácidos, o lo que es lo mismo, el número de veces que aparece dicho aminoácido en las secuencias codificantes de nuestra bacteria. Para calcular el RSCU alcanza con dividir el número de ocurrencias de cada codón entre el total de veces que se utiliza el aminoácido y multiplicarlo por el número de codones que tiene el aminoácido (\(n_i=6\) en el caso de Arginina, \(n_i=4\) en el caso de Valina, ver el código genético).
Por lo tanto, utilizando la ecuación (1.7), tenemos que por ejemplo, para el codón CGU de la Arginina su RSCU es igual a
\[\begin{equation} RSCU_{R,CGU}=\frac{x_{CGU}}{\frac{1}{6}\sum_{j=1}^6\ x_{R,j}}=6 \times \frac{666}{16294}=0,2452 \end{equation}\]
Para el codón CGC la cuenta es igual a
\[\begin{equation} RSCU_{R,CGC}=\frac{x_{CGC}}{\frac{1}{6}\sum_{j=1}^6\ x_{R,j}}=6 \times \frac{1320}{16294}=0,4861 \end{equation}\]
y así sucesivamente con todos los codones de este aminoácido, lo que nos deja con una tabla como la siguiente:
| Codón | Conteo | RSCU |
|---|---|---|
| \(\textbf{CGU}\) | \(666\) | \(0,2452\) |
| \(\textbf{CGC}\) | \(1320\) | \(0,4861\) |
| \(\textbf{CGA}\) | \(1457\) | \(0,5365\) |
| \(\textbf{CGG}\) | \(1818\) | \(0,6694\) |
| \(\textbf{AGA}\) | \(5913\) | \(2,1774\) |
| \(\textbf{AGG}\) | \(5120\) | \(1.8854\) |
| \(\textbf{Suma}\) | \(16294\) | \(6\) |
De la misma manera, para la Valina, el RSCU del primer codón es igual a
\[\begin{equation} RSCU_{V,GUU}=\frac{x_{GUU}}{\frac{1}{4}\sum_{j=1}^4\ x_{V,j}}=4 \times \frac{7123}{18605}=1,5314 \end{equation}\]
mientras que para el resto de los codones se calcula en forma análoga, hasta obtener la siguiente tabla:
| Codón | Conteo | RSCU |
|---|---|---|
| \(\textbf{GUU}\) | \(7123\) | \(1,5314\) |
| \(\textbf{GUC}\) | \(2144\) | \(0,4610\) |
| \(\textbf{GUA}\) | \(1125\) | \(0,2419\) |
| \(\textbf{GUG}\) | \(8213\) | \(1,7658\) |
| \(\textbf{Suma}\) | \(18605\) | \(4\) |
En ambos aminoácidos se observa que el uso de codones se aparta bastante de la hipótesis de uso similar. Mientras que en el primer caso (Arginina) los codones favorecidos son los codificados por el dueto (del sexteto), es decir AGA y AGG, en el segundo caso (Valina) los codones favorecidos son los que terminan en U o en G.
1.2.1 Exploración multivariada: Análisis de Correspondencia
Este método fue desarrollado por Benzecrí (Benzecri 1980), originalmente para trabajar con tablas de frecuencias. En resumen, la idea subyacente es dada una tabla de \(n \times p\) (casos x variables) encontrar un sistema de \(k\) ejes ortogonales (con \(k < min(n,p)\)) que condense el máximo de variación en los datos originales. Esto es posible en la medida en que las variables (casos) estén correlacionados en alguna medida y cuanto mayor sea esa asociación más varianza será condensada en pocos ejes (dimensiones). La métrica adoptada es la de la distancia \(\chi^2\), o sea, para la celda correspondiente a la fila \(i\) y columna \(j\), la distancia será
\[\begin{equation} d_{ij}=(O_{ij}-E_{ij})^2/E_{ij} \tag{1.9} \end{equation}\]
donde \(O_{ij}\) es el número observado y \(E_{ij}\) el número esperado para esa celda, usualmente el número total de observaciones multiplicado por las frecuencias relativas marginales en la tabla.
Aplicando el COA a una tabla de frecuencias observadas vamos a obtener una serie de valores propios y vectores propios asociados (“eigen-values” y “eigen-vectors”), tantos como el mínimo de Filas-1 y Columnas-1. Cada uno de los valores propios está relacionado con la varianza explicada por la nueva dimensión correspondiente. La suma de todos los valores propios es equivalente al 100% de la varianza por lo que cada uno explicará una fracción correspondiente al valor propio sobre la suma de todos (la traza de la matriz de valores propios). A su vez, cada uno explica menor varianza que el anterior (están ordenados por el procedimiento de extracción). El punto importante es determinar con cuántos ejes (nuevas dimensiones) nos vamos a quedar. Cuantas más dimensiones retengamos mayor cantidad de la varianza original retendremos, pero al costo de dificultar la interpretación (el objetivo del COA es poder capturar la esencia de los datos, cuantas más dimensiones más difícil de visualizar la situación). Existen varios métodos para determinar el número de dimensiones a retener, pero en última instancia esto es una materia subjetiva. Un enfoque es gráfico (“scriplot”) y consiste en encontrar el cambio en la pendiente de los valores propios graficados en función de su número ordinal.
Otra aproximación es retener los valores propios hasta que expliquen menos de lo que se esperaría por azar si cada unas de las variables originales explicara la misma proporción de la varianza. Por ejemplo, en una tabla de uso de aminoácidos, dado que hay 20 AAs la varianza promedio explicada por cada uno es 5% y por lo tanto luego de un COA en AAs retendríamos todos aquellos valores propios que expliquen más de 5% de la varianza. Cada una de las observaciones originales tendrá ahora unas nuevas coordenadas en el nuevo sistema de ejes ortogonales (las dimensiones). O sea, si por ejemplo retuvimos de nuestro COA en uso de AAs 3 dimensiones (combinaciones de las 20 originales) cada gen podrá representarse ahora en un espacio tridimensional. Asimismo, las variables originales están representadas de distinta manera en las nuevas dimensiones, es decir tienen distinta participación en cada una de ellas. Esto apunta a una de las características más destacables del COA, su simetría Filas/Columnas lo que permite el auxilio de representar en forma conjunta (en el mismo gráfico) la distribución de observaciones y variables (genes y AAs, por ejemplo) aunque las distancias entre observaciones y variables deben tomarse con cautela.
La clave del éxito con el COA es lograr “etiquetar” las nuevas dimensiones con factores entendibles, biológicamente significativos, que vayan más allá de las propias variables medidas. Ejemplo de esto en genómica podría ser la expresión diferencial, los compartimientos celulares, proteínas IMP (“integral membrane proteins”), etc, cuando se correlacionan con alguno de los ejes del análisis. La forma tradicional de trabajo, al menos en el campo de la genómica comparativa, consiste en determinar cuántos valores propios retener para luego graficarlos de a pares intentando ver la distribución conjunta de observaciones y variables en las nuevas dimensiones. Se puede además intentar correlacionar variables externas con las nuevas dimensiones, buscando “etiquetas” para las dimensiones. Un link interesante de análisis de correspondencia: http://www.micheloud.com/FXM/COR/index.htm
1.2.2 Composición de proteínas
Hasta ahora hemos centrado nuestros análisis composicionales en bases nucleotídicas y codones, pero la mayor parte de las secuencias procarióticas terminan siendo traducidas en proteínas. Por lo tanto, vamos a dedicar un breve espacio a tratar esta temática. En muchos casos los archivos de partida para un análisis bioinformático (en cualquiera de los formatos estándar, como FASTA o GenBank) solo incluyen la secuencia nucleotídica, dado que a partir de la misma es posible deducir directamente la secuencia de AAs (si conocemos el código genético correspondiente).
Existen muchos programas para realizar la traducción de estas secuencias, pero el transeq (de la suite EMBOSS) es muy flexible y eficiente, aceptando diversos formatos de entrada. Dentro del EMBOSS también tenemos toda una serie de programas para facilitar el análisis de secuencias proteicas. Entre ellos (hay muchos más): antigenic para encontrar sitios antigénicos, charge que hace un diagrama de carga, digest reporta sobre enzimas proteolíticas, emowse para buscar en proteínas por peso molecular de fragmentos de digestión, fuzzpro para buscar patrones, helixturnhelix para identificar motivos de unión a ácidos nucléicos, iep para calcular el punto isoeléctrico, makeprotseq para crear secuencias aleatorias, pepcoil para predecir regiones “coiled-coil”, pepstats para calcular diversos estadísticos de proteínas, preg para buscar expresiones regulares en proteínas, pepwindow para realizar un ploteo de hidropatía, tmap para predecir segmentos trans-membrana, psiphi para calcular ángulos torsionales y patmatmotifs que escanea una proteína con motivos del PROSITE (http://prosite.expasy.org/).
Diferencias en propiedades de los AAs
Existen 22 aminoácidos proteinogénicos o naturales (los que se incorporan directamente en las proteínas). De entre ellos, 20 son codificados directamente, mientras que la selenocisteína y la pirrolisina se codifican a partir de modificaciones en la lectura de codones stop (con símbolos IUPAC/IUBMB Sec o U, y Pyl u O, respectivamente). Como se puede apreciar en la figura 1.4, los 20 aminoácidos codificados directamente (que suelen ser los más abundantes) poseen importantes diferencias en las propiedades físico-químicas, lo que lleva a agruparlos de diferentes maneras, de acuerdo al grupo de propiedades de relevancia.
Típicamente, podemos distinguir:
- i) Polares/hidrofílicos - N, Q, S, T, K, R, H, D, E, (C, Y)16,
- ii) No-polares / hidrofóbicos - (G), A, V, L, I, P, Y, F, W, M, C,
- iii) enlace de H - C, W, N, Q, S, T, Y, K, R, H, D, E,
- iv) contienen azufre - C, M,
- v) con carga negativa a pH neutro / acídicos - D, E, (C),
- v) con carga positiva a pH neutro / básicos - K, R, (H),
- vi) Ionizables - D, E, H, C, Y, K, R,
- vii) Aromáticos - F, W, Y, (H, pero sin absorción significativa de UV), viii ) Alifáticos - G , A, V, L, I, P,
- ix) Forman enlaces covalentes cruzados (puentes di-sulfuro) - C,
- x) Cíclicos - P
(ver por ejemplo http://www.mcb.ucdavis.edu/courses/bis102/AAProp.html).
Figura 1.4: Diagrama de Venn con las propiedades de los diferentes aminoácidos. Figura tomada de Wikipedia (CC BY-SA 3.0), versión en castellano modificada a partir del archivo “Amino Acids Venn Diagram (es).svg”
Las diferencias en las propiedades físico-químicas llevan a importantes diferencias en la estructura de las proteínas y hacen que los cambios observados (substituciones) entre proteínas homólogas (descendientes de un ancestro común) tengan distribuciones particulares, las que se suelen representar en matrices de substituciones17 (ver Figura 1.5). Estas diferencias, por lo tanto, tendrán un gran impacto en muchos de los procesos evolutivos, así como en la reconstrucción de los mismos. De hecho, algunos AAs poseen bastante similaridad estructural y fisicoquímica con otros, por ejemplo Phe con Tyr e Ile con Val, lo que hace que los mismos sean más propensos a intercambiarse cuando las presiones mutacionales son suficiéntemente fuertes.
Figura 1.5: Matriz de sustitución aminoacídica BLOSUM62. Las letras en filas y columnas de la matriz refieren a los veinte aminoácidos naturales codificados directamente, y su color refleja sus tipos estructurales y fisicoquímicos. Las entradas numéricas de la matriz son proporcionales al logaritmo de la frecuencia con la que se observa la sustitución de un aminoácido por otro en proteinas homólogas. Valores positivos denotan mayores frecuencias de sustitución; lo contrario ocurre para valores negativos. Figura tomada de Wikipedia (CC BY-SA 3.0), versión original del archivo “Blosum62-dayhoff-ordering.svg”
Por ejemplo, mientras que en el caso de Phe con Tyr no hay diferencias en el contenido GC de los codones implicados, en el segundo caso (Ile con Val) se trata del cambio A\(\leftrightarrow\)G en primera posición del codón y por lo tanto estará sujeto a cambio cuando el sesgo mutacional sea hacia GC (AT). Otros casos de cambios relativamente frecuentes incluyen Asp con Glu, Glu con Gln, Ile con Leu, Arg con Lys, Leu con Met, His con Tyr y Trp con Tyr. De todos estos solo el último implica un cambio de dos bases, el resto de solo una base y casi todos independientes del contenido GC (de todos estos solo His con Tyr implica cambio de GC). Esto podría estar relacionado con un fuerte proceso selectivo para la optimización de la estructura del código genético (Novozhilov, Wolf, and Koonin 2007), aunque existen teorías alternativas.
Bajando un poco más el nivel de conservación (pero aún con “score” positivo en la matriz BLOSUM62) tenemos varios cambios que requieren cambio de contenido GC en los codones implicados.
Un recurso extremadamente útil para comparar las propiedades de los aminoácidos, así como para trabajar a partir de las mismas, es la “AAindex database” (http://www.genome.jp/aaindex/) (Kawashima, Ogata, and Kanehisa 2000). En la misma se encuentra registrada información para unas 500 propiedades diferentes, así como información adicional acerca de los autores, la publicación original y otras auxiliares. La posibilidad de acceder a estas propiedades a través de la biblioteca seqinr de R facilita enormemente la exploración de hipótesis más complejas que se relacionen con la substitución de AAs (ver por ejemplo (L. Spangenberg et al. 2011)).
Diferencias de composición entre clases de proteínas
Existen diferentes clases de proteínas de acuerdo al criterio que se utilice para clasificarlas y, en general, de acuerdo al criterio usado poseen una composición diferente. Por ejemplo, de acuerdo a la forma podemos clasificarlas en “Fibrosas”, “Globulares” y “Mixtas”. Las fibrosas presentan largas cadenas polipeptídicas, estructura secundaria atípica, son insolubles en agua y en disoluciones acuosas (v.gr., queratina, colágeno y fibrina). Las globulares pliegan sus cadenas en una forma esférica apretada, con los grupos hidrófobos hacia el interior y grupos hidrófilos hacia afuera. Esto hace las mismas sean solubles en disolventes polares como el agua. Entre ellas se encuentran los anticuerpos, las enzimas, algunas hormonas y proteínas de transporte. Finalmente, las mixtas poseen una parte fibrilar y otra parte globular.
Las proteínas solubles en agua suelen poseer los residuos hidrofóbicos (Leu, Ile, Val, Phe y Trp) escondidos en el medio de la proteína (en su estructura), mientras que los hidrofílicos se encuentran expuestos en la superficie. Al mismo tiempo, las proteínas de membrana (IMPs) suelen contar con anillos externos hidrofóbicos, los cuales sirven de anclaje en la bicapa lipídica. Por otro lado, proteínas que deben interactuar con moléculas de carga positiva suelen tener residuos de carga negativa en su superficie (Glu y Asp) y viceversa (Arg y Lys). Algunos AAs poseen propiedades muy particulares, con un efecto sustancial en la estructura de la proteína. Pro por ejemplo, que forma codos en la estructura y Cys, que forma puentes di-sulfuro estabilizando estructuras.
Un grupo de particular interés son las proteínas de membrana, que juegan un papel muy importante en el intercambio de moléculas entre la célula y el exterior, así como en la transducción de señales. Por esta razón es muy importante poder predecir qué secuencias se corresponden a IMPs. Existen básicamente dos tipos de IMPs, las de \(\alpha\)-hélices (TMH) y las de barriles-\(\beta\) (TMB). Las primeras se encuentran en la membranas internas bacterianas y en las membranas plasmáticas bacterianas, aunque a veces también en membrana externa. Es la mayor categoría de proteínas transmembrana y una parte importante de todas las proteínas humanas. Las proteínas de conformación tipo barriles-\(\beta\) se encuentran solo en membrana externa de bacterias Gram-negativas, pared celular de Gram-positivas y membrana externa de organelos.
Aunque existen distintos algoritmos y programas, la predicción de la pertenencia a alguna de estas dos clases se basa esencialmente en el mismo paradigma, básicamente el alineamiento a otras proteínas o estructuras conocidas (Bigelow and Rost 2009). Un concepto clave en este sentido es el de “transferencia de homología a través de alineamientos”. Bajo este paradigma, dos proteínas homólogas poseerán un alineamiento razonable. Por lo tanto, es posible transferir información derivada de evidencia experimental de una proteína a otra de interés para la que no se posee información. Claramente la capacidad de extender las conclusiones depende de la calidad del alineamiento. Ejemplo de esto serían regiones transmembrana en proteínas TMH: existen métodos específicos para la predicción de ambas clases de IMPs. Los métodos para detección de TMHs se basan en la presencia de una o más hélices transmembrana. Dentro de estos métodos se encuentra TMHMM (http://www.cbs.dtu.dk/services/TMHMM/).
Entre los programas que permiten discriminar entre proteínas TMB o no-TMB se encuentra TMB-Hunt, que utiliza un algoritmo k-Nearest Neighbour (k-NN) sobre la composición aminoacídica de las proteínas. El mismo permite además incluir pesos diferenciales para los aminoácidos, así como información evolutiva, lo que consigue una precisión mayor al 92% con una sensibilidad mayor al 91% (Garrow, Agnew, and Westhead 2005). Finalmente, una alternativa interesante es utilizar InterScanPro (http://www.ebi.ac.uk/Tools/pfa/iprscan/), una herramienta que combina la información de hasta 14 programas de predicción diferentes. Los programas que se pueden seleccionar a través de esta herramienta son: BlastProDom (busca en la base de dominios ProDom), FPrintScan (busca en la base PRINTS de grupos de motivos), HMMPIR (busca en modelos HMM de la Protein Sequence Database, PSD), HMMPfam (busca en modelos HMM de la base PFAM), HMMSmart (busca en modelos HMM de la base SMART), HMMTigr (busca en modelos HMM de la base TIGRFAMs), ProfileScan (busca contra perfiles de PROSITE), HAMAP (busca contra perfiles de HAMAP), PatternScan (nueva versión de búsqueda sobre PROSITE), SuperFamily, SignalPHMM, TMHMM, HMMPanther y Gene3D. El costo computacional de estas búsquedas es muy elevado, y por lo tanto se debe ser muy cauteloso a la hora de enviar varios trabajos simultáneamente.
Ejemplo 1.4
Escherichia coli es una bacteria común en el intestino de los mamíferos. En la misma, el regulón del fosfato comprende un conjunto de más de 20 genes no-ligados cuyos promotores pueden ser regulados simultáneamente por controles superpuestos y separados. En particular, Los genes phoS, phoA y phoE son inducidos por la privación de fosfato y están sujetos a un control positivo por el producto del gen phoB y a un control negativo por el producto del gen phoR. Por otra parte, el sistema de transporte específico de fosfato (Pst) está codificado por cuatro genes estrechamente relacionados entre sí y situados juntos: phoS, phoT, pstA y pstB.
La siguiente es la secuencia de aminoácidos correspondientes al gen pstA, un transportador integral de membrana (Accesion K01992 del GenBank):
“MAMVEMQTTAALAESRRKMQARRRLKNRIALTLSMATMAFGLFWLI WILMSTITRGIDGMSLALFTEMTPPPNTEGGGLANALAGSGLLILW ATVFGTPLGIMAGIYLAEYGRKSWLAEVIRFINDILLSAPSIVVGL FVYTIVVAQMEHFSGWAGVIALALLQVPIVIRTTENMLKLVPYSLR EAAYALGTPKWKMISAITLKASVSGIMTGILLAIARIAGETAPLLF TALSNQFWSTDMMQPIANLPVTIFKFAMSPFAEWQQLAWAGVLIIT LCVLLLNILARVVFAKNKHG”
Si realizamos el análisis composicional de la misma llegamos la siguiente tabla:
| A | C | D | E | F | G | H | I | K | L | M | N | P | Q | R | S | T | V | W | Y |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 38 | 1 | 3 | 11 | 13 | 21 | 2 | 27 | 10 | 41 | 17 | 9 | 12 | 8 | 13 | 16 | 22 | 18 | 9 | 5 |
El largo total es de \(296\) aminoácidos. La suma de los 4 aminoácidos hidrofóbicos más importantes (A, I, L y V) es de \(38+27+41+18=124\), es decir, \(\frac{124}{296}=41,9\%\) del total de aminoácidos. Si bien la comparación tiene poco sentido si no conocemos la composición global del genoma (es decir, del resto de las proteínas del genoma), sin duda es una buena pista de que se trata de una proteína de membrana (donde los aminoácidos hidrofóbicos juegan un papel relevante debido a la naturaleza lipídica de la bicapa que forma la membrana).
PARA RECORDAR
- A nivel genómico la evolución se da por un conjunto de eventos como las mutaciones puntuales, las duplicaciones de genes y regiones o las inserciones y “deleciones” de secuencias nucleotídicas. El estudio de las variaciones en la composición de los genomas es lo que se puede llamar genómica composicional y forma parte de la genómica comparativa.
- La idea del análisis de correspondencia es que dada una tabla de \(n \times p\) (casos x variables) encontrar un sistema de \(k\) ejes ortogonales (con \(k < min(n,p)\)) que condense el máximo de variación en los datos originales. Esto es posible en la medida en que las variables (casos) estén correlacionados en alguna medida y cuanto mayor sea esa asociación más varianza será condensada en pocos ejes (dimensiones).
- La métrica adoptada en el COA es la de la distancia \(\chi^2\), o sea, para la celda correspondiente a la fila \(i\) y columna \(j\), la distancia será \(d_{ij}=(O_{ij}-E_{ij})^2/E_{ij}\) donde \(O_{ij}\) es el número observado y \(E_{ij}\) el número esperado para esa celda, usualmente el número total de observaciones multiplicado por las frecuencias relativas marginales en la tabla. El punto importante es determinar con cuantos ejes (nuevas dimensiones) nos vamos a quedar. Cuantas más dimensiones retengamos mayor cantidad de la varianza original retendremos, pero al costo de dificultar la interpretación.
- Desde el punto de vista biológico, lo relevante del COA, además de identificar si podemos condensar la varianza en pocos ejes, es poder etiquetar cada eje con uno o unos pocos factores biológicos relevantes, tanto sean composicionales, de estructura del código, ambientales, etc.
- Los distintos aminoácidos poseen diferentes propiedades que permiten agruparlos en diversos grupos, de acuerdo a las mismas. Por ejemplo, tenemos aminoácidos polares, apolares, hidrofílicos, hidrofóbicos, pequeños, medianos y grandes, alifáticos y aromáticos, etc. La importancia de esto es que permiten generar proteínas con diferentes propiedades y estructura tridimensional.
- La redundancia en el código genético permite que un mismo aminoácido sea codificado por diferentes codones. Entre otras cosas, eso permite una mayor tolerancia a las mutaciones, ya que mutaciones que cambian el codon pero no el aminoácido no suelen tener mayor impacto en la estructura de la proteína (aunque puede ocurrir esto último debido a diferencias en la velocidad de traducción y su impacto en el plegamiento de la proteína).
- El contenido G+C de los genomas es una variable extremadamente importante ya que se encuentra directamente asociada, entre otras cosas, a la composición de las proteínas. Diferentes hipótesis se han propuesto para explicar la enorme variabilidad entre los procariotas (\(20\%-80\%\)), pero en esencia se dividen en neutralistas y seleccionistas.
1.3 Genómica comparativa
Como vimos más arriba, solo es posible entender la biología de los
organismos si entendemos los procesos evolutivos subyacentes
(Dobzhansky 1964). De hecho, pese a que nuestra visión antropocéntrica
nos incline a vernos sustancialmente diferente de otras especies, desde
el punto de vista genómico compartimos una enorme similaridad con otras
especies. A nivel genómico la similitud nucleotídica es mayor al 98% en
algunos casos, como por ejemplo con el chimpancé (Pan troglodytes)
(Sequencing and Consortium 2005). Las diferencias que observamos entre las
especies pueden, en general, correlacionarse con diferencias a nivel
genómico, tanto por la presencia/ausencia de genes así como diferencias
en las secuencias que producen proteínas con diferencias
estructurales/funcionales o modificaciones en las redes regulatorias. La
genómica comparativa trata precisamente de identificar diferencias en
los genomas, entender los procesos evolutivos que llevan a esas
diferencias y en algunos casos de correlacionar estas diferencias con el
fenotipo o aún el ambiente donde se desarrollan los organismos.
Comparación de secuencias génicas
La base de la genómica comparativa se encuentra en la determinación de homología en secuencias, es decir la inferencia sobre el origen ancestral compartido de las mismas. En especial, la determinación de la relación de ortología (ancestría común por especiación, ver nota al pie más arriba) juega un papel central en la genómica comparativa ya que suele implicar (aunque no siempre) la realización de la misma función en diferentes organismos. La determinación de ortología es un problema usualmente complejo, particularmente cuando hay varias especies implicadas. La base de este proceso es la identificación de secuencias, en todas las especies de interés, que presenten un nivel de similaridad tal que permita inferir la relación de ortología. Debido a que es usualmente necesario explorar muchos pares (solo la comparación de dos bacterias requiere del orden de \(10^7\) comparaciones), se requiere de herramientas que identifiquen similaridad rápidamente, lo que lleva al tipo de alineamientos heurísticos como el del programa BLAST.
Usualmente las secuencias ortólogas son las más similares en las otras especies. Esto ha llevado al criterio práctico del Reciprocal Best Hit (RBH): un par de secuencias se consideran ortólogas si cada una de ellas es la mejor “selección” (la más similar) en su correspondiente genoma, de la otra. Un criterio de este tipo es necesario para evitar relaciones confusas debido a la existencia de parálogos18. Cuando se comparan dos especies este criterio está suficientemente bien definido; sin embargo, cuando se trata de más de dos especies la existencia de parálogos en alguna de ellas puede llevar a identificar distintas secuencias como pertenecientes al grupo de ortólogas en dicha especie de acuerdo a qué pares de especies se tomen en cuenta.
Este problema nos lleva a otros dos criterios de utilidad práctica a la hora de identificar ortólogos. Por un lado, cuando la similaridad entre secuencias es debida a eventos de especiación (definición de ortólogos, ver más arriba) el orden de los distintos genes dentro del genoma será relativamente similar durante mucho tiempo. Esto se debe a que los re-arreglos genómicos suelen tener un importante costo evolutivo; en procariotas en particular existen estructuras llamadas operones, que facilitan la expresión coordinada de un conjunto de genes cuyos productos interactúan directamente y que tienden a conservarse en la evolución. Este ordenamiento puede ser usado para determinar las relaciones de ortología, ya que además del alto nivel de identidad de las secuencias podemos requerir que los genes en el vecindario de ambas especies sean los mismos y estén ordenados de similar manera. Todo esto es altamente dependiente de la distancia evolutiva entre los organismos considerados, ya que a mayor tiempo de divergencia menos conservación de estos patrones podemos esperar.
La otra aproximación, conocida como filogenómica, implica una cuidadosa reconstrucción filogenética19, a partir de la cual se intenta conciliar los resultados del alineamientos de las secuencias homólogas. Con un poco de suerte es posible determinar correctamente las relaciones de ortología y paralogía de nuestras secuencias. Los resultados obtenidos son altamente dependientes de la reconstrucción filogenética, por lo que es necesario tener mucho cuidado en la selección de secuencias para realizar la misma, en la correcta selección del modelo de substitución (tanto aminoácidica como nucleotídica) y el algoritmo de reconstrucción filogenética. En la medida de lo posible, es bueno explorar la sensibilidad del análisis frente a posibles cambios en el proceso (por ejemplo la selección de otro método de reconstrucción).
Existe una gran cantidad de software específico para atacar el problema de la detección y determinación de ortólogos, así como bases de datos para distintos grupos filogenéticos. Entre estas últimas podemos destacar COG (Cluster of Orthologous Groups, http://www.ncbi.nlm.nih.gov/COG/), una base de datos construida a partir de RBH con consistencia en al menos tres linajes para procariotas y su extensión eucariótica KOG, así como EGO (http://compbio.dfci.harvard.edu/tgi/ego/) una base de ortólogos para eucariotas. Entre los programas podemos mencionar Inparanoid (http://inparanoid.sbc.su.se/) que consiste en RBH seguido de agrupamiento de in-parálogos (parálogos dentro de una especie), así como OrthoMCL (http://www.orthomcl.org/) que a diferencia del primero usa E-value y porcentaje de identidad para asignar las proteínas a diferentes agrupamientos. Finalmente, un programa muy interesantes es Orthostrapper (http://sonnhammer.sbc.su.se/download/software/), y su visualizador OrthoGUI, que permite obtener valores de soporte estadístico para la ortología de cada par de secuencias en un alineamiento.
Selección positiva
Otro ejemplo interesante de las posibilidades de la genómica comparativa es la determinación de sitios en el genoma (o genes) que se encuentran evolucionando bajo selección positiva, o dicho en otras palabras, donde la selección se orienta hacia una adaptación a un ambiente particular (mejora en el “fitness”). Existen diversas formas de estudiar el efecto de la selección a nivel molecular, pero uno de los usualmente más aceptados es el de la relación \(\frac{K_a}{K_s}\) (también conocida como \(\omega\), o dN/dS). Esta es la relación entre el número de substituciones no-sinónimas por sitio no-sinónimo con respecto al número de substituciones sinónimas por sitio sinónimo. Como las substituciones sinónimas es de esperar que sean encontradas a una tasa usualmente mucho más alta que las no-sinónimas (ya que no generan cambios aminoacídicos y por lo tanto no repercuten en la función proteica), aquellos sitios con una relación \(\frac{K_a}{K_s} > 1\) se consideran bajo selección positiva. El problema principal radica en determinar cuales apartamientos del 1 resultan estadísticamente significativos.
Existen diferentes formas de estimar \(K_a\) y \(K_s\), los más complejos usando estimación por máxima-verosimilitud20, pero un método aproximado suele dar resultados equivalentes cuando los datos son suficientes. En esencia, en el método aproximado, a partir de un alineamiento de secuencias, se realiza el conteo de sustituciones sinónimas y no-sinónimas, con corrección por substituciones múltiples, para luego calcular la relación \(\frac{K_a}{K_s}\). Es importante tener en cuenta que el alineamiento debe realizarse a nivel de aminoácidos, para luego realizar el traducción reversa a nucleótidos, asegurándose de esta forma que la comparación se realiza codón a codón. Dentro del software clásico para estimar esta relación podemos mencionar PAML (http://abacus.gene.ucl.ac.uk/software/paml.html) que realiza la estimación por máxima-verosimilitud, mientras que entre las fáciles de usar podemos mencionar la función KaKs de la biblioteca seqinr de R.
Vías
Las proteínas suelen realizar sus funciones particulares como partes de procesos más complejos, que envuelven otras proteínas, en diferentes etapas de esos procesos. El conjunto de proteínas (u otras móleculas) que participan en un determinado proceso se conocen como vía. A modo de ejemplo podemos recordar la vía de la pentosas fosfato, durante la cual se utiliza la glucosa para generar ribosa y poder reductor a través de NADPH. Las distintas vías existentes en cada organismo se constituyen en el “mapa” de los distintos procesos biológicos que cada organismo es capaz de realizar. Si bien el número de organismos donde todos los procesos bioquímicos son conocidos al detalle es relativamente escaso, la genómica comparativa es una alternativa importante para determinar la capacidad de realización de distintos procesos en otros organismos.
La idea central en esta estrategia consiste en ubicar en el genoma de interés ortólogos de los genes que constituyen una vía en un organismo modelo, como puede ser E. coli. Asumiendo que los genes ortólogos posean aún la misma función, la completitud de la vía (es decir, la presencia en el organismo de interés de todos los genes que la constituyen en el modelo) permite inferir la capacidad de realizar el proceso bioquímicos en el organismo de interés. Existen diferentes bases de datos con representaciones de las diferentes vías en varios organismos, pero probablemente la más conocida es KEGG Pathway (http://www.genome.jp/kegg/pathway.html). La misma permite la búsqueda por diferentes criterios, posee información de varios organismos, muestra la información en forma gráfica y existe una API (Application Programming Interface) que facilita el acceso a los datos a través de software.
Por otro lado, la existencia de información de las diferentes vías puede ser usada durante el proceso de anotación genómica, ya que si conocemos que un organismo realiza determinado proceso bioquímico entonces deben estar presentes todos los genes que constituyen esa vía. Si bien en algunos organismos las vías pueden estar formadas por diferentes genes, en general es posible determinar la falta de genes claves en las distintas vías, lo que usualmente se debe a errores en el proceso de anotación o a cambios desconocidos en las vías (esto último de gran interés científico).
PARA RECORDAR
- La genómica comparativa trata de identificar diferencias en los genomas, entender los procesos evolutivos que llevan a esas diferencias y en algunos casos de correlacionar estas diferencias con el fenotipo o aún el ambiente donde se desarrollan los organismos.
- La base de la genómica comparativa se encuentra en la determinación de homología en secuencias, es decir la inferencia sobre el origen ancestral compartido de las mismas. En especial, la determinación de la relación de ortología (ancestría común por especiación, ver más arriba) juega un papel central en la genómica comparativa ya que suele implicar (aunque no siempre) la realización de la misma función en diferentes organismos. Usualmente las secuencias ortólogas son las más similares en las otras especies y esto ha llevado al criterio práctico del Reciprocal Best Hit (RBH), es decir, un par de secuencias se consideran ortólogas si cada una de ellas es la mejor “selección” (la más similar) en su correspondiente genoma, de la otra. Cuando la similaridad entre secuencias es debida a eventos de especiación el orden de los distintos genes dentro del genoma será relativamente similara durante mucho tiempo.
- La aproximación filogenómica, implica una cuidadosa reconstrucción filogenética, a partir de la cual se intenta conciliar los resultados del alineamientos de las secuencias homólogas. Los resultados obtenidos son altamente dependientes de la reconstrucción filogenética, por lo que es necesario tener mucho cuidado en la selección de secuencias para realizar la misma, en la correcta selección del modelo de substitución (tanto aminoácidica como nucleotídica) y el algoritmo de reconstrucción filogenética.
- Existen diversas formas de estudiar el efecto de la selección a nivel molecular, pero uno de los usualmente más aceptados es el de la relación \(K_a/K_s\), que es la relación entre el número de substituciones no-sinónimas por sitio no-sinónimo con respecto al número de substituciones sinónimas por sitio sinónimo. Como las substituciones sinónimas es de esperar que sean encontradas a una tasa usualmente mucho más alta que las no-sinónimas, aquellos sitios con una relación \(K_a/K_s\) por encima de 1 se consideran bajo selección positiva. El problema principal radica en determinar cuales apartamientos del 1 resultan estadísticamente significativos.
1.4 Genómica funcional
Las proteínas son los principales efectores a nivel celular (e.g., las enzimas), así como también juegan un rol fundamental en las estructuras celulares y las diferentes maquinarias, por lo que si intentamos entender el funcionamiento de células y tejidos a este nivel es necesario conocer cuándo, cuánto y dónde se producen. Por ejemplo, si queremos ver las diferencias entre un tejido determinado en estado normal y en el mismo tejido pero con un tumor, podríamos medir el nivel de cada proteína en ambos tejidos y comparar luego los niveles correspondientes para entender las diferencias. Desafortunadamente, la medición de proteínas a escala masiva (lo que se conoce como proteómica) es un tema aún relativamente en pañales, si bien existen cientos de estudios desarrollados a esta escala en diferentes especies. Se trata de técnicas caras y complejas, que no han experimentado aún una explosión similar a la que se experimentó a nivel de la medición de ácidos nucleicos.
De acuerdo a una versión esquemática del paradigma de la biología molecular (paradigma que imperó durante muchos años), la información se almacena en el ADN, la que luego es transcrita hacia el ARN y que luego se traduce en proteínas. Si juntamos esto con el hecho de que es relativamente sencillo medir cantidades de ARN en forma masiva (en realidad ADNc), la idea de utilizar la medición de los transcritos como un proxy a la medición de proteínas surge de inmediato. Es decir, si suponemos que la tasa de producción de proteínas es directamente proporcional a la tasa de transcripción, entonces medir la cantidad de transcritos es una buena y fiable indicación de la producción de proteínas (aunque existe una compresión de rango entre el logaritmo de la relación de proteínas y el logaritmo de la relación de mensajeros, como lo muestran L. Spangenberg et al. (2013)). Claramente, a pesar de su enorme importancia en el desarrollo de la disciplina, esta visión simplificada del paradigma de la biología molecular ha sido ampliamente sobrepasada por la información generada en los últimos años, incluyendo los descubrimientos de los microARNs y de un gran número de otros ARNs que juegan un papel fundamental en la regulación de la expresión génica. Más aún, la vida media de los diferentes transcritos no es necesariamente igual, lo que conlleva un error a la hora de utilizar su cantidad (no su tasa de producción) como indicador de la producción de proteínas.
A pesar de todos estos problemas, el análisis de la expresión de los ARN mensajeros (ARNm) sigue siendo la principal herramienta para entender los cambios a nivel de expresión en los distintos tipos de órganos y tejidos, así como en células bacterianas o de otras especies. En un principio, los microarrays fueron fundamentales para esta tarea ya que permitían interrogar miles de genes al mismo tiempo. Desde los originales spot arrays que eran impresos por un robot cartesiano mediante un bloque de agujas que depositaban las sondas en una placa de vidrio, diferentes tecnologías se fueron desarrollando. Una de las principales limitaciones de los primeros arrays era la necesidad de utilizar algún tipo de “referencia”, es decir algo contra lo que contrastar las mediciones debido a las imperfecciones de las impresiones con agujas. Posiblemente de las tecnologías más conocida es la desarrollada por la empresa Affymetrix (fundada por el bioquímico uruguayo Alejandro Zaffaroni) que utilizando la misma tecnología que se utiliza para la fabricación de chips de semiconductores (esencialmente la fotolitografía) consiguieron desarrollar arrays de altísima densidad. La figura 1.6 muestras dos arrays de esta marca, a la izquierda un chip para genoma humano, mientras que a la derecha aparece un chip para genoma de ratón; en la parte inferior aparece un fósforo a efectos de imaginar el tamaño de los mismos. La principal ventaja de estos arrays respecto a los típicos spotted era la muy alta precisión con los que eran fabricados, lo que permitía evitar tener que usar una referencia. Sin embargo, el principal problema con estos arrays era el costo de desarrollo asociado a cada nuevo diseño, por lo que quedaron reducidos a las pocas especies que eran modelo para la investigación (humano, ratón, Arabidopsis thaliana, etc.).

Figura 1.6: Dos chips de Affymetrix, uno para genoma humano a la izquierda, el otro para genoma de ratón. El fósforo debajo es solo a fines de comparación del tamaño. La fecha de expiración de ambos es en el año 2006, por lo que su uso seguramente fue anterior (Schutz, trabajo propio (based on copyright claims)., CC BY 2.5, https://commons.wikimedia.org/w/index.php?curid=694717).
Un poco más tarde, la tecnología de la empresa Agilent, la impresión laser de los arrays permitió combinar una altísima precisión con la posibilidad de fabricar los mismos a gusto del consumidor y sin costos adicionales, lo que llevó a utilizarlos en decenas de nuevas especies. En efecto, en lugar de utilizar tinta de diferentes colores, el array se construía imprimiendo un nucleótido por vez en cada posición de una sonda, utilizando como tintas 4 soluciones con los nucleótidos A, C, G y T. La precisión de esta tecnología permitía realizar análisis en experimentos de un solo canal o en experimentos con dos canales, lo que en ciertos casos posibilitaba reducir los costos (por ejemplo, cuando existe una referencia natural para cada caso, utilizando el mismo array se elimina la necesidad de dos arrays).
Finalmente, en años recientes, las tecnologías de secuenciación masiva llegaron a este campo y mediante la secuenciación del ARNc permiten obtener una representación mucho más amplia y fidedigna de las proporciones de los distintos mensajeros. Además, como se obtienen las secuencias de los mismos, es posible identificar variantes (que son a su vez producto de variantes en el ADN) asociadas a cambios en la expresión entre individuos, lo que se conoce como eQTL (“expression Quantitative Trait Locus”). La principal diferencia entre el análisis de expresión con arrays respecto a la secuenciación (que se conoce como RNA-seq) es que en el primer caso la señal obtenida es analógica (un número en una escala continua), mientras que en el segundo caso es digital (en realidad el conteo de los reads que mapean a un transcrito particular).
La idea de las comparaciones es similar en las diferentes tecnologías, aunque cada tecnología requiere de detalles específicos a la hora de analizar los datos. En general, si vamos a comparar el perfil de expresión en dos tratamientos diferentes, por ejemplo tejido sano y tumoral, requerimos de varios individuos en cada tratamiento a fin de tener en cuenta la variación entre individuos y de que nuestras conclusiones se puedan extrapolar de alguna manera a la población general. Luego de haber obtenido los perfiles de expresión en cada individuo necesitamos realizar algún tipo de normalización, ya que las cantidades absolutas suelen depender de la preparación de las bibliotecas. Luego de normalizadas las muestras debemos comparar gen a gen, lo que suele hacerse mediante un modelo lineal sobre los datos transformados de alguna manera. En los casos en que se compara un tratamiento contra una referencia, se acostumbra a utilizar la distribución del logaritmo de la relación de cambio entre las dos condiciones (que se llama \(logFC\)). Es decir, si la expresión del gen \(i\) en el tratamiento es \(mRNA_{i,1}\) y en la referencia o control es \(mRNA_{i,2}\), entonces
\[\begin{equation} logFC_i=log\left[\frac{mRNA_{i,1}}{mRNA_{i,2}}\right] \tag{1.10} \end{equation}\]
Las ventajas de esta transformación son varias. Por un lado, tanto las distribuciones de conteo (en el caso de RNA-seq) como las de señal luminosa tienen una cola larga a la derecha, es decir, una serie de valores muy altos y alejados de donde se concentra la mayor parte de la distribución; el logaritmo “corrige” de alguna forma eso, transformándolas en distribuciones parecidas a la normal. En segundo caso, mientras que la relación \(\frac{mRNA_{i,1}}{mRNA_{i,2}}\) es asimétrica (cuando el numerador es más pequeño que el denominador la relación queda entre 0 y 1, mientras que al revés va de 1 a \(+\infty\)), la relación transformada \(log\left[\frac{mRNA_{i,1}}{mRNA_{i,2}}\right]\) es simétrica y centrada en cero, además de relativamente normal. Con esta transformación, por ejemplo si hay solo dos grupos a comparar, la comparación podría ser a través de un test de \(t\) (la distribución). Como realizamos miles de comparaciones a la vez (una por gen), la significación estadística de cada gen debe ajustarse para múltiples ensayos, lo que usualmente se realiza a través de algún método como Benjamini-Hochberg.
Estudios epigenéticos y epigenómicos
Hasta ahora hemos manejado los aspectos de la genética que tienen que ver con la transmisión de información como algo bastante rígido y donde hay poco margen para cambiar lo recibido de los progenitores. Sin embargo, existen una serie de mecanismos que tienen que ver con la regulación que pueden transmitirse de padres a hijos y a su vez ser modificados posteriormente de recibidos. El estudio de estos mecanismos particulares es el objetivo de la epigenética y de la epigenómica. Básicamente, la epigenética se centra en los procesos que regulan la activación y desactivación de genes, tanto en lo que hace a cómo y cuándo se produce esto. Por otra parte, la epigenómica se refiere a los cambios globales que ocurren a nivel de genoma que permiten activar o desactivar un conjunto de genes, tanto a nivel de células como de organismos.
En general, los procesos epigenéticos son los responsables de controlar el crecimiento y desarrollo de un organismo. En situaciones particulares, la alteración del control puede llevar a procesos patogénicos, como por ejemplo el cáncer. Lo trascendente de este proceso es que factores ambientales pueden ser los causantes del desajuste en la regulación y por lo tanto en el desarrollo de patologías. Dentro de los factores ambientales conocidos, la exposición a determinadas sustancias químicas aparece como muy relevante, aunque también la dieta parece estar relacionada con alteraciones en la regulación. Además, existe cierta evidencia de los cambios en la regulación podrían ser transmisibles.
Existen dos formas en las que el epigenoma es capaz de marcar el ADN para la regulación génica: a) la metilación y b) la modificación de las histonas. En la primera, grupos metilo se intercalan en la molécula de ADN modificando la función cuando esto ocurre en un gen promotor. En general, la metilación del ADN actúa reprimiendo la transcripción génica. En mamíferos entre el \(60\%\) y el \(90\%\) de todas las CpG están metiladas. Por otra parte, en la segunda forma de acción una serie de modificaciones en las histonas (acetilación, metilación, desmetilación, fosforilación, ubiquitilación, etc.) modifican el arrollamiento del ADN y por lo tanto la disponibilidad para la transcripción.
Por supuesto, existen arrays disponibles para el estudio de las modificaciones epigenómicas, así como métodos para realizar los estudios a través de secuenciación masiva.
PARA RECORDAR
- Las proteínas son los principales efectores a nivel celular, así como también juegan un rol fundamental en las estructuras celulares y las diferentes maquinarias, sin embargo, desafortunadamente, la medición de proteínas a escala masiva (lo que se conoce como proteómica) es un tema aún relativamente nuevo: se trata de técnicas ´aún caras y complejas y que no han experimentado aún una explosión similar a la que se experimentó a nivel de la medición de ácidos nucléicos.
- Como sabemos, la información se almacena en el ADN, la que luego es transcrita hacia el ARN y que luego se traduce en proteínas. Si a su vez consideramos el hecho de que es relativamente sencillo medir cantidades de ARN en forma masiva (en realidad ADNc), la idea de utilizar la medición de los transcritos como un proxy a la medición de proteínas surge de inmediato. Es decir, si suponemos que la tasa de producción de proteínas es directamente proporcional a la tasa de transcripción, entonces medir la cantidad de transcritos es una buena y fiable indicación de la producción de proteínas.
1.5 Conclusión
El estudio de la vida se ha nutrido a lo largo de los siglos del avance tecnológico; ejemplos claros de esto son la aparición del microscopio (que permitió vislumbrar que la vida habitaba lugares insospechados en formas diminutas) o los avances de la química, que permitieron comprender los mecanismos subyacentes al funcionamiento de la maquinaria celular y la herencia desde una perspectiva molecular. El uso de herramientas matemáticas también ha sido fundamental para el desarrollo teórico de la biología: pensemos en la importancia que tuvo la simple aproximación de Mendel al estudio de la herencia de caracteres.
No es de extrañar que en décadas marcadas por el avance de las ciencias de la informática y del procesamiento tecnológico de compuestos químicos también exista una nueva vigorización de nuestro entendimiento de la biología. En este capítulo presentamos varias de las áreas de estudio que han surgido o se han visto vitalizadas por la aparición de métodos de secuenciación masiva (i.e., las distintas áreas de la genómica), discutiendo su impacto en la tipificación y estudio de la diversidad genética, la identificación y análisis evolutivo de organismos o el análisis de la expresión génica y uso de codones.
En el resto del libro trataremos dos áreas sumamente importantes de la genética: el estudio de la herencia a nivel poblacional y el análisis cuantitativo. Las tecnologías y la forma de análisis que vimos en este capítulo reaparecerán constantemente a medida que aprendamos cómo se observa la genética a través de estas dos potentes lentes conceptuales. Antes de seguir con ese recorrido, es importante remarcar nuevamente que estas dos áreas surgieron mucho antes que las tecnologías que mencionamos en este capítulo: una vez más en la historia de la biología, la aparición de tecnologías da nuevos datos y perspectivas de análisis a marcos teóricos que ya existían, ayudando a testear hipótesis, ampliar nuestro conocimiento e impulsando nuevas preguntas sobre el fenómeno de la herencia.
1.6 Actividades
1.6.1 Control de lectura
- Existen muchas maneras de medir la diversidad biológica. Mencione al menos una, explicitando la forma de cálculo y en qué contexto puede ser útil la medida elegida.
- ¿Qué es un microarray? Explique la utilidad de esta herramienta, poniendo algún ejemplo de su aplicación.
- En las últimas décadas se han desarrollado un conjunto de disciplinas que permiten la obtención y análisis masivo de datos biológicos. Identifique el objeto de estudio de las siguientes:
- Genómica
- Transcriptómica
- Proteómica
- Metagenómica
- Metatranscriptómica
- Epigenómica
- Actualmente se acepta que todos los organismos provenimos de un ancestro común (denominado LUCA). ¿Cómo se explica entonces que existan diferencias tan grandes entre los organismos vivos? Mencione al menos una aproximación para el estudio de esta diversidad biológica.
- Defina el concepto de codón. ¿Todos los organismos usan el mismo conjunto de codones? ¿Los codones son todos usados en igual proporción?
1.6.2 ¿Verdadero o falso?
- Un codón solo codifica para un aminoácido, pero un aminoácido puede estar codificado por más de un codón.
- La estructura del código genético refleja las propiedades fisicoquímicas de los aminoácidos codificados en el llamado “código genético universal”.
- La secuenciación y comparación de secuencias de ARNr 16S permite la identificación y estudio filogenético de todos los organismos vivos.
- El contenido GC de los organismos no es uniforme, existiendo variaciones a lo largo del árbol de la vida.
- Todos los cambios genéticos heredables se encuentran codificados en el genoma de los organismos, por lo que secuenciando y comparando secuencias genéticas podemos comprender en su totalidad el proceso de herencia de información entre individuos (y en las poblaciones).
Solución
- Verdadero. Este concepto suele denominarse “redundancia” o “degeneración” del código genético. Entre otras cosas, eso permite una mayor tolerancia a las mutaciones, ya que mutaciones que cambian el codon pero no el aminoácido no suelen tener mayor impacto en la estructura de la proteína (aunque puede ocurrir esto último debido a diferencias en la velocidad de traducción y su impacto en el plegamiento de la proteína).
- Verdadero. Los cambios sinónimos suelen llevar con más frecuencia a aminoácidos de características fisicoquímicas similares, hecho que podría adjudicarse a un régimen selectivo que favoreció el evitar que el proceso mutacional lleve a cambios radicales en las proteínas codificadas.
- Falso Si bien es cierto que la secuenciación de ARN ribosomal es útil para identificar tanto a organismos procariotas como eucariotas, es importante recordar que las subunidades secuenciadas varían en los dominios de la vida: en procariotas si se recurre al estudio de la subunidad 16S, mientras que en eucariotas se recurre al 18S (recordar que no existe subunidad 16S en eucariotas).
- Verdadero. El contenido de GC (o lo que es lo mismo, AT) varía ampliamente entre organismos. Ya en la década de 1960 se determinó que existen bacterias cuyo genoma se encuentra en un rango que va de 25 a 75% de contenido GC. Tal es la diferencia, que en muchos casos es posible identificar transferencias de material genético entre diferentes especies bacterias observando que tramos del genoma desvían fuertemente de la tendencia general observada en el resto del genoma (se ahondará en este tema en el capítulo Genética de Poblaciones Microbianas).
- Falso. Si bien este fue un paradigma dominante durante décadas, la existencia de transmisión de información epigenética (e.g. a través de patrones de metilación y acetilación de histonas) muestra que es posible la herencia de información biológica que no se encuentra codificada a nivel nucleótidico. Disciplinas como la epigenómica intentan estudiar la relevancia de estos procesos en la herencia de los individuos e incluso la evolución.
Ejercicios
 Ejercicio 1.1
Ejercicio 1.1
Dada la siguiente secuencia “AUGGGAUUUCCGCCCAUUUACGUG”
a. Determinar de que ácido nucléico se trata (ADN o ARN). b. Si tiene sentido traducirla a una secuencia de aminoácidos tomandola como hebra codificante y en marco de referencia. c. Escribir en sentido 5’–>3’ la hebra complementaria pero en ADN (hebra molde) d) calcular el contenido G+C.
Solución
a. Se trata de ARN ya que entre sus bases aparece Uracilo en lugar de Timina.
b. En principio parece tener sentido, por lo que asumiendo que está en marco de referencia correcto y y que se trata de la hebra codificante, separamos en codones “AUG GGA UUU CCG CCC AUU UAC GUG”, que utilizando el código genético se traduce como MGFPPIYV.
c. “CAC GTA AAT GGG CGG AAA TCC CAT”.
d. 6G + 6C= 12 G o C en un total de [8 codones x 3 bases c/u = 24 bases], por lo tanto 12/24=0.5 o 50%.
Ejercicio 1.2
Un investigador obtuvo la siguiente secuencia (según él) de un nuevo gen que sería responsable de la generación de radiactividad por parte de una especie bacteriana que lo posee (presentamos la secuencia ordenada en tripletes y en marco de lectura correcto, para mayor facilidad de visualización):
GTC TCT CGT GAT GCG GTT GTC ATA AGA GCT GGC TCT GAG CTG
AAT TGA AGC ATA TCC TTC GCA GTC TTA AAA GGG TTG GGA TTA
TCA ACC TCC CAC GCA ATA CCG GCG TTC AAC AGT TTA CCA TCA
TCT ACT GAC GCG TGA TGT TGC GGA GCG ACG GGA AGT GAT
AAA AGT TAC TTT ACALa especie bacteriana de la que supuestamente fue aislado el gen, posee un contenido GC del 80 %.
Por otra parte, un estudiante de su grupo afirma que no solamente no se trata de la secuencia de un nuevo gen, sino que no se trata de la secuencia de ningún gen. Más aún, plantea que esta secuencia fue generada en forma aleatoria por una máquina y que posiblemente el investigador ha sido engañado por alguien. El conteo de bases dio lo siguiente:
\[ \begin{cases} A: 46 \\ C: 40 \\ G: 45 \\ T: 49 \end{cases} \]
mientras que el conteo de aminoácidos fue el siguiente:
\[ \begin{cases} STOP: 2 \\ A: 7 \\ C: 2 \\ D: 3 \\ E: 1 \\ F: 3 \\ G: 5 \\ H: 1 \\ I: 3 \\ K: 2 \\ L: 5 \\ N: 2 \\ P: 2 \\ R: 2 \\ S: 11 \\ T: 4 \\ V: 4 \\ Y: 1 \end{cases} \]
a. ¿Quién de los dos parece tener razón? Argumenta las razones que te llevan a esa conclusión.
b. ¿Cuántas cisteínas hay codificadas en la secuencia y dónde se encuentran?
c. ¿Cuántas serinas y cuántas argininas serían de esperar en una secuencia de este largo si se tratase de una secuencia creada al azar y cuántas fueron observadas? ¿Y la suma de las dos (es decir, para los codones de los sextetos)?
d. Determinar mediante un test estadístico si el número de Serinas y de Argininas observadas cae dentro de lo esperado, o si debemos rechazar dicha hipótesis nula.
Solución
a. Algunos hechos hacen sospechar que no se trata de una secuencia presente en un organismo vivo tal y como los conocemos:
- El codón de iniciación no es ATG ni tampoco ninguna de las formas alternativas más usuales (esta argumento no es excluyente, pero es raro de observar).
- En la posición 46, es decir el inicio del codón 16, corresponde con el codón TGA que es un codón terminación, lo que nos dejaría con una proteína de 15 aminoácidos de largo. Lo mismo ocurre en la posición 139, es decir el inicio del codón 47, por lo que tampoco parece mucho mejor la situación del fragmento interno entre ambos codones STOP.
- La secuencia no termina en ninguno de los codones STOP conocidos.
- La distribución de la frecuencia de bases no se aparta mucho de lo esperado para una secuencia obtenida al azar (\(\frac{180}{4} = 45\)). En cambio, si el contenido del genoma bacteriano fuese de \(80\%\), entonces esperaríamos \(180∗0,80 2 = 72\) bases para G y lo mismo para C, mientras que esperaríamos \(180∗0,20 2 = 18\) bases para A y lo mismo para T.
b. Dos cisteínas, y se encuentran codificadas por los codones 48 y 49.
c. La serina está codificada por 6 codones y Arginina por otros 6 codones, de los 64 que hay en el código genético, o lo que es lo mismo, de \(4^3 = 64\) posibles combinaciones de cada bases (A, C, G, T) en 3 posiciones. Como hay un total de 60 codones en la secuencia, esperamos: \(\text{Ser} = 60 ∗ \frac{6}{64} = 5,625\), \(\text{Arg} = 60 ∗ \frac{6}{64} = 5,625\). Se observaron 11 y 2 respectivamente, lo que en ambos casos parece algo alejado del esperado. Sin embargo, la suma de ambos sextetos da 13, mientras que el esperado es \(\text{Ser}+\text{Arg} = 60 ∗ \frac{12}{64} = 11,25\), lo que parece mucho más cercano.
d. Un test de chi-cuadrado de “goodness-of-fit” (bondad de ajuste) podría ser adecuados en este caso.
Ejercicio 1.3
Observando la tabla con el código genético universal agrupar los aminoácidos por el número de codones que los codifican y resumir el número de aminoácidos con cada multiplicidad. ¿Qué característica comparten la mayor parte de los duetos (los aminoácidos codificados por dos codones)?
Solución
- 1 codon: Met, Trp
- 2 codones: Phe, Tyr, His, Gln, Asn, Lys, Asp, Glu, Cys
- 3 codones: Ile
- 4 codones: Val, Pro, Thr, Ala, Gly
- 6 codones: Leu, Arg, Ser
La característica que comparten 7 de 9 duetos es que se encuentran en la tercer columna de la tabla del código, es decir, tienen una A en segunda posición del codon.
Ejercicio 1.4
En un planeta de otra galaxia existe vida con algunas similitudes a la que existe en nuestro planeta. En particular, también las proteínas parecen ser la base estructural y funcional de la vida, mientras que los ácidos nucléicos son los responsables del mantenimiento y duplicación de la información. Sin embargo, a diferencia de nuestro planeta, los aminoácidos proteinogénicos codificados son 24, mientras que las bases nucleótidicas análogas a las de nuestro ADN son 5. Suponiendo que los mecanismos moleculares en dicho planeta han sido seleccionados para ser altamente eficientes
a. ¿De qué largo mínimo deberían ser los codones?
b. Si el código genético en ese planeta tuviese algún nivel de redundancia (más de un codón para el mismo aminoácido), ¿de qué largo mínimo serían los codones? En ese caso, ¿cuál sería el número promedio de codones por aminoácido?
c. ¿Cómo se compara esa redundancia promedio con la existente en nuestro planeta?
d. ¿Qué ventaja potencial tiene la redundancia en el código?
Solución
a. Si existen 5 bases nucleotídicas y 24 aminoácidos, entonces con codones de largo 2 tenemos \(5 \times 5 = 25\) combinaciones posibles. Si consideramos la existencia de un codón STOP, entonces el mínimos es \(24 + 1 = 25\) codones, por lo que alcanza con largo de codón 2.
b. Si queremos algo de redundancia vamos a precisar un código genético con largo mayor a 2. El siguiente largo es 3, por lo que ahora tenemos \(5 \times 5 \times 5 = 5^3 = 125\) codones. Como hay \(24\) AAs \(+1\) STOP \(= 25\) “necesidades”, entonces la redundancia promedio será igual a \(\frac{125}{25} = 5\).
c. En nuestro planeta tenemos 20 aminoácidos proteinogénicos codificados directamente, por lo que equivale a \(20\) AAs \(+1\) STOP = 21 “necesidades”. Sin embargo, al existir solo 4 bases nucleotídicas en nuestro código y el largo de nuestros codones ser de 3, entonces tenemos un total de \(4 \times 4 \times 4 = 4^3 = 64\) posibilidades. Por lo tanto, en nuestro planeta, la redundancia promedio es de \(\frac{64}{21} \approx 3,05\).
d. La redundancia en el código tiene como ventaja que es capaz de absorber determinadas mutaciones (cambio de bases) sin cambiar el aminoácido codificado, lo que lo hace un sistema con cierta robustez o tolerancia a los fallos en la duplicación de la información (mutaciones).
Ejercicio 1.5
Asumiendo que existen 20 aminoácidos (los estándar),
a. ¿Cuántas proteínas diferentes de largo 5 aminoácidos sería posible tener?
b. Cuántas serían posibles de largo \(n\)?
c. Considerando que una proteína de largo promedio en procariotas es del orden de los 300 aminoácidos, ¿cuántas proteínas diferentes, con ese largo se podrían codificar?
Solución
a. Se podrían codificar \(20 \times 20 \times 20 \times 20 \times 20 = 20^5 = 3,200,000\).
b. De largo \(n\) se pueden codificar \(20^n\).
c. En el caso de \(n = 300\), tenemos \(20^300 \approx \infty\) para los fines prácticos
Bibliografía
Se denomina como epigenético todo cambio heredable que no tiene como base un cambio a nivel de nucleótido en la secuencia de ADN. A modo de ejemplo, cambios en el estado de metilación de un nucleótido pueden influir en el estado de compactación de la cromatina y así en la expresión de algunos genes; esto puede resultar en cambios fenotípicos heredables, aún cuando en ningún momento se haya reemplazado un nucleótido por otro.↩︎
En ecología se define a una comunidad como un conjunto de poblaciones de dos o más especies que conviven al mismo tiempo en una misma área geográfica. Un ecosistema está compuesto de una comunidad y del entorno abiótico que comprende a la misma. Ambos conceptos son amplios, y pueden referir a unidades ecológicas de tamaños y características diversas.↩︎
Diferentes individuos tenderán a reproducirse con mayor o menor éxito debido a una variedad de factores tales como la capacidad de sobrevida en un entorno dado o la fecundidad, entre otros. En la teoría evolutiva, este concepto se ve representado por el término fitness, que refiere a la contribución reproductiva de un individuo a su próxima generación. Así, diremos que individuos con un mayor fitness tienden a dejar mayor descendencia en la próxima generación. Algunos autores hacen referencia al fitness de un individuo, mientras que otros refieren al fitness de un genotipo particular o incluso de un alelo.↩︎
Interesantemente, la metagenómica ha permitido el descubrimiento reciente de una diversidad arqueana antes desconocidas, lo cual ha llevado a una fuerte controversia en esta área de la filogenética contemporánea. En particular, el descubrimiento en 2015 de un filo arqueano denominado Asgard y el análisis en conjunto de diferentes marcadores moleculares ha aportado evidencia congruente con una “hipótesis de dos dominios” para el árbol de la vida. Según esta hipótesis el dominio Eukarya sería en realidad un subgrupo del dominio Arquea, fuertemente emparentado con el filo Asgard. Más allá de qué hipótesis posea mayor respaldo, esta controversia ilustra hasta qué punto nuevas evidencias provenientes de la genómica y la metagenómica aportan a debates de primer nivel en la teoría biológica; otros ejemplos de relevancia teórica y práctica son mencionados más adelante en esta misma sección.↩︎
En teoría evolutiva se dice que dos caracteres son ortólogos si derivan de un único ancestro común a dos especies. A modo de ejemplo, los genes codificantes para mioglobina de humano y chimpancé son ortólogos, ya que se infiere que el ancestro común a ambas especies poseía una unica copia para este gen; una vez se dió el evento de especiación, pasó a existir una copia para cada especie.↩︎
Técnica analítica que permite determinar con alta precisión qué moléculas orgánicas y/o biomoléculas se encuentran presentes en una muestra.↩︎
El contenido GC se calcula usualmente como \(GC=(N_C+N_G)/(N_A+N_C+N_G+N_T)\), donde \(N_A\)...\(N_T\) corresponde al número de veces que aparece cada base en una secuencia.↩︎
Hay 6 de estas combinaciones, pero como la frecuencia de las dos elegidas más la de las otras dos debe sumar 1, basta con calcular la mitad; v. gr., GC=1-AT.↩︎
Los orbitales de los anillos aromáticos de las bases nitrogendas de los nucleótidos que componen la molécula de ADN interaccionan a medida que se apilan en el polímero. Esta interacción no covalente es un factor fundamental que aumenta la estabilidad de la doble hélice de ADN.↩︎
Si bien el código genético se encuentra ampliamente conservado en los tres dominios de la vida, este claramente no es universal. Mitocondrias, cloroplastos y algunos protozoarios unicelulares o bacterias del género Mycoplasma, entre otros, emplean códigos genéticos con variaciones en algunos codones.↩︎
El \(GC_{skew}\) se calcula tomando una ventana de tamaño determinado y de acuerdo con la siguiente expresión: \(GC_{skew}=(N_G-N_C)/(N_G+N_C)\).↩︎
figuran entre paréntesis cuando entran en la categoría parcialmente↩︎
Las matrices de sustituciones aminoacídicas son matrices que reflejan la frecuencia evolutiva de sustitución de pares de aminoácidos. Existen diferentes variantes (e.g. matrices PAM, matriz BLOSUM62), dependiendo del set de proteínas y las asunciones con las que las mismas fueron construídas. Estas matrices juegan un rol clave en la implementación de algoritmos de alineamiento de proteínas, como puede ser el empleado por el programa BLAST mencionado anteriormente.↩︎
Se dice que dos secuencias son parálogas si son producto de una duplicación ancestral. Un caso clásico para ilustrar este concepto es el de la familia de las hemoglobinas. Existen genes codificantes para \(\alpha\) y \(\beta\)-hemoglobina en humano y chimpancé, y se ha inferido que estos genes también existían en el ancestro común a ambas especies. Los genes de \(\alpha\)-hemoglobina y los de \(\beta\)-hemoglobina de las especies son ortólogas entre sí. Sin embargo, diríamos que la \(\alpha\)-hemoglobina de humano es paráloga a la \(\beta\)-hemoglobina de chimpancé, ya que su “parentesco” no se debe al proceso de especiación, si no a la duplicación del gen codificante para la hemoglobina ancestral.↩︎
Una filogenia representa la historia evolutiva inferida para un conjunto de diferentes entidades, incluyendo patrones de ancestría y las tasas de cambio que llevaron a la diversidad observada. Si bien el ejemplo más paradigmático puede ser el de una filogenia de especies (donde se representa la evolución del grupo) también es posible pensar en una filogenia de genes o cualquier otra entidad autoreplicante que se modifique a lo largo del tiempo.↩︎
Dado un modelo probabilístico, la estimación de parámetros por máxima-verosimilitud es un método que busca estimar los valores que maximizan la posibilidad de observar un conjunto de datos dado según el modelo.↩︎